I'm referring to the authors of the Pradhan et al. paper simply as
Pradhan for the sake of convenience, even though the paper indicated
that there were three authors who had an equal contribution to the
paper, who were Prashant Pradhan, Ashutosh Kumar Pandey, and Akhilesh
Mishra. [https://
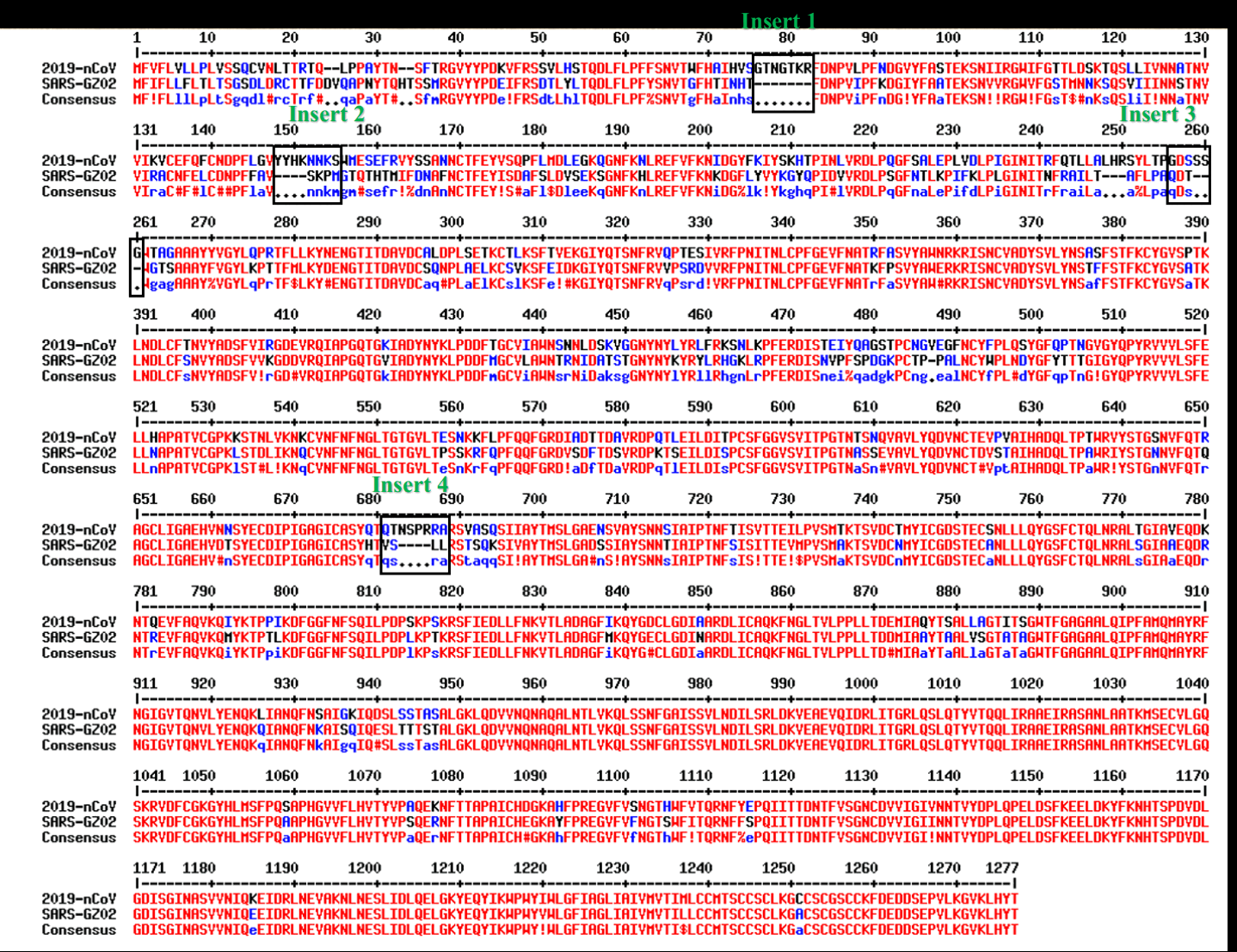
Pradhan did a pairwise alignment of the spike protein of SARS-CoV-2 against SARS-CoV, and he identified 4 regions where SARS-CoV had gaps in the alignment, which he interpreted to be inserts in SARS-CoV-2 relative to SARS-CoV:
When Pradhan did BLAST searches for the so-called inserts and the surrounding amino acids, he found that for example the last 6 aa of the first 7 aa insert had a perfect match to an HIV sequence from Thailand, and the last 2 aa of the second insert along with the next 4 aa had a perfect match to an HIV sequence from Kenya:
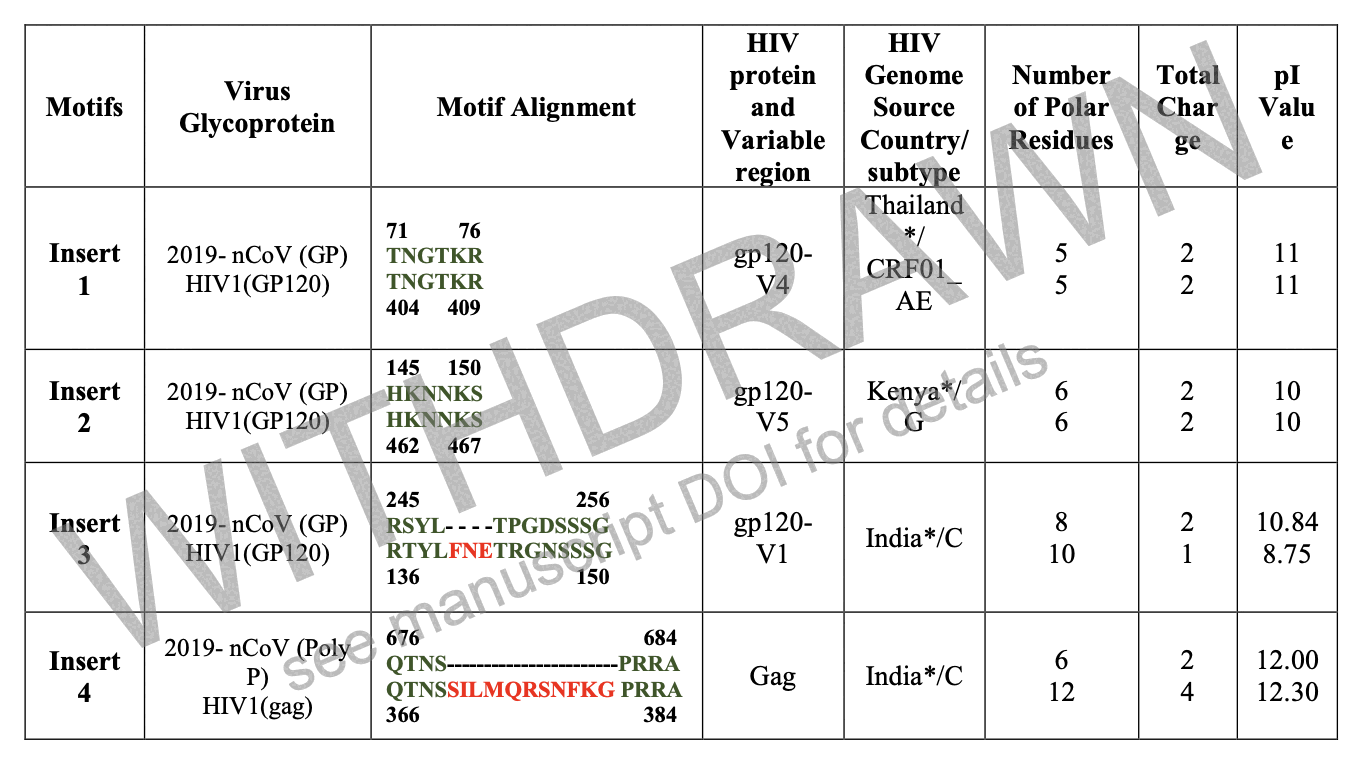
However all of the matches were either so short or they had so many mismatches that the matches were highly likely to occur by chance. In BLAST, the E-value indicates how many similarly close or closer matches are expected to occur by chance for the given combination of query sequence and target sequences, but Pradhan deceptively did not report the E-values of his matches anywhere.
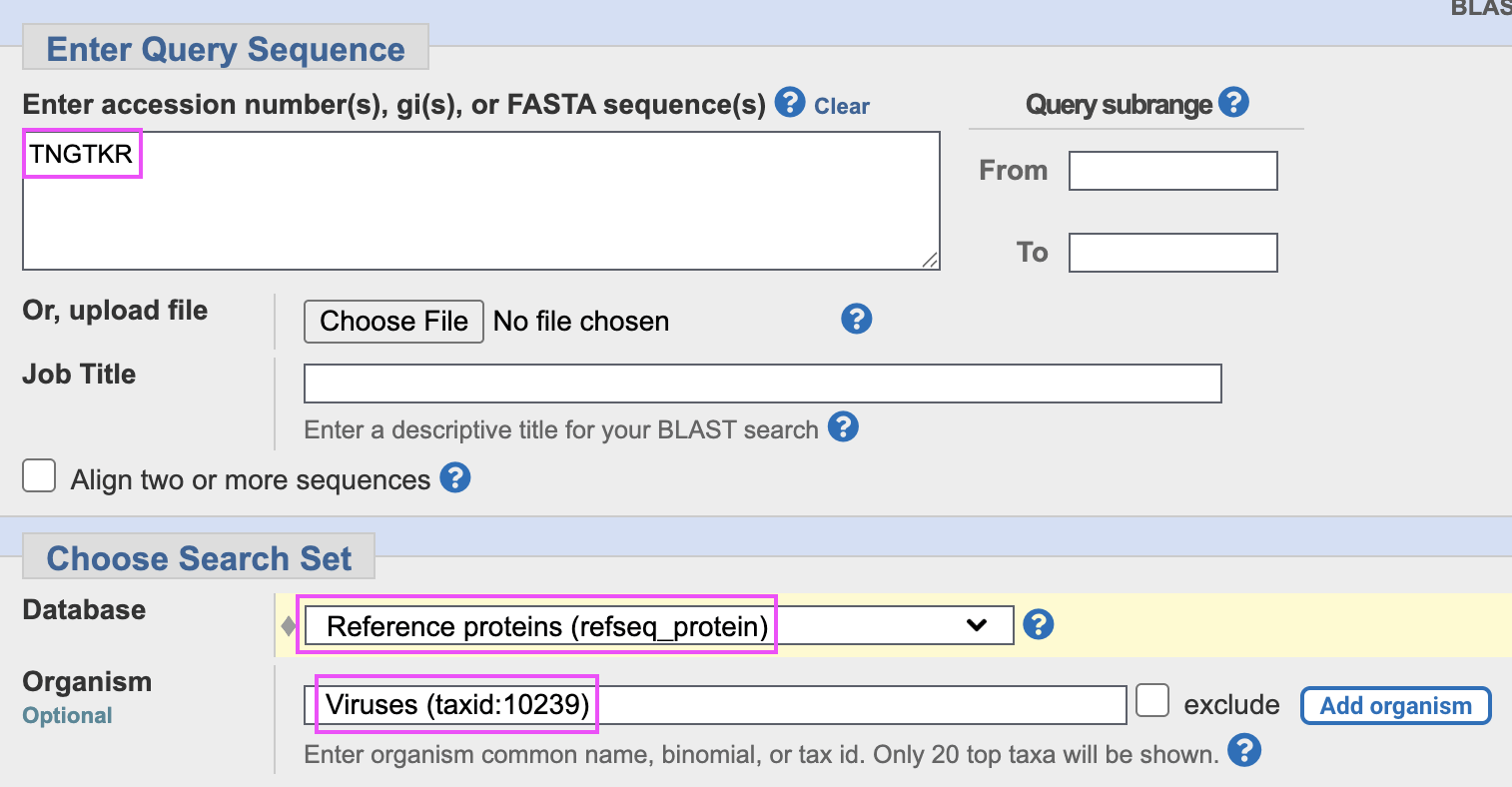
I went to the web interface for protein BLAST:
https://TNGTKR, which was the match Pradhan found to
the first so-called insert, I set organism to "Viruses (taxid:10239)",
I clicked "Add organism", and I set the
second organism to "Betacoronavirus pandemicum (taxid:3418604)" and I clicked the exclude
checkbox next to it. Then under "Algorithm
parameters" I increased "Max target
sequences" to 1000, and I clicked "BLAST". Pradhan's match occured in a set of 10
similar sequences from Thailand whose accesions start with
AFU28. The E-values of the matches were 428, which means
that about 428 similarly close or closer matches were expected to occur
by chance:
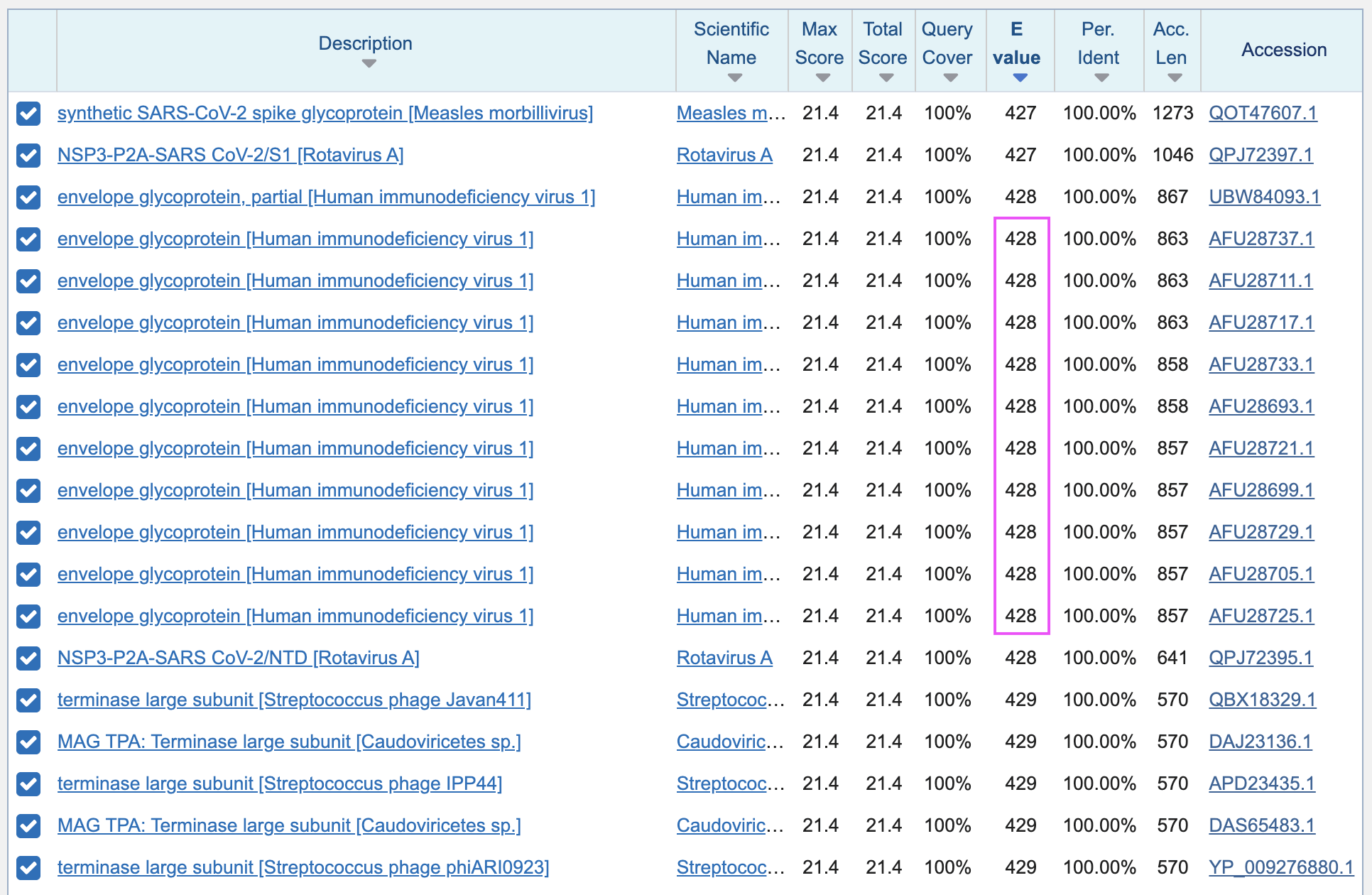
There was a total of 310 sequences with a perfect 6 aa match to the
query TNGTKR, so the E-value of 428 was fairly close to the
actual number of perfect matches. The perfect matches occurred among
species like "Acute bee paralysis virus",
"Cnidium virus 2", "Echovirus E11", "Barkedji
virus", "Wild boar ephemerovirus",
"Rotavirus A", "phage
csAssE-Sib", "environmental Halophage
eHP-31", "bajarodmic virus 944", "Wuhan House Fly Virus 1", "Vibrio phage PERU2", and so on. Pradhan's matches
to the other so-called inserts also occurred among many other species of
viruses besides HIV.
When I did similar BLAST searches for Pradhan's matches to the other so-called inserts, all of the matches had an E-value above 100:
| Insert | SARS-CoV-2 | HIV | E-value | Results with same or lower E-value |
|---|---|---|---|---|
| 1 |
TNGTKR
|
TNGTKR
|
428 | 310 |
| 2 |
HKNNKS
|
HKNNKS
|
212 | 8 |
| 3 |
RSYL---
|
RSYLFNETRGNSSSG
|
281 | 236 |
| 4 |
QTNS-----------
|
QTNSSILMQRSNFKGPRRA
|
705 | 75 |
In March 2026 when I did my BLAST searches, the nr
protein BLAST database was about 4 times bigger than in January 2020
when Pradhan did his BLAST searches, so my E-values were also about 4
times bigger than the E-values in January 2020. However when Pradhan
identified the matches to the so-called inserts, he searched for the
inserts and an unspecified number of surrounding residues from either
side of the insert, so there were multiple possible sequences that could
match the region around each insert. So in reality his E-values were
likely even higher than the values in my table above (unless for example he performed separate searches for
multiple different 6 aa segments around the second insert, and not for a
single longer segment that consisted of the second insert and an
arbitrary number of surrounding residues on either side, but even in
that case, the total E-value of all separate searches added together
would likely be more than 4 times higher than the value for a single 6
aa search).
The nr database contains a large number of sequences
from widely studied species of viruses like SARS-CoV-2, HIV-1, and
influenza A. But in order to restrict a BLAST search to a database that
only has one or a few representative samples for each species of virus,
I searched for TNGTKR in the refseq_
database instead, so that I restricted the organism to viruses. I got an
E-value of about 36 for the perfect matches, so even within a fairly
small pool of proteins from virus refseqs, the E-values for perfect
matches to the 6 aa segment were still very high:
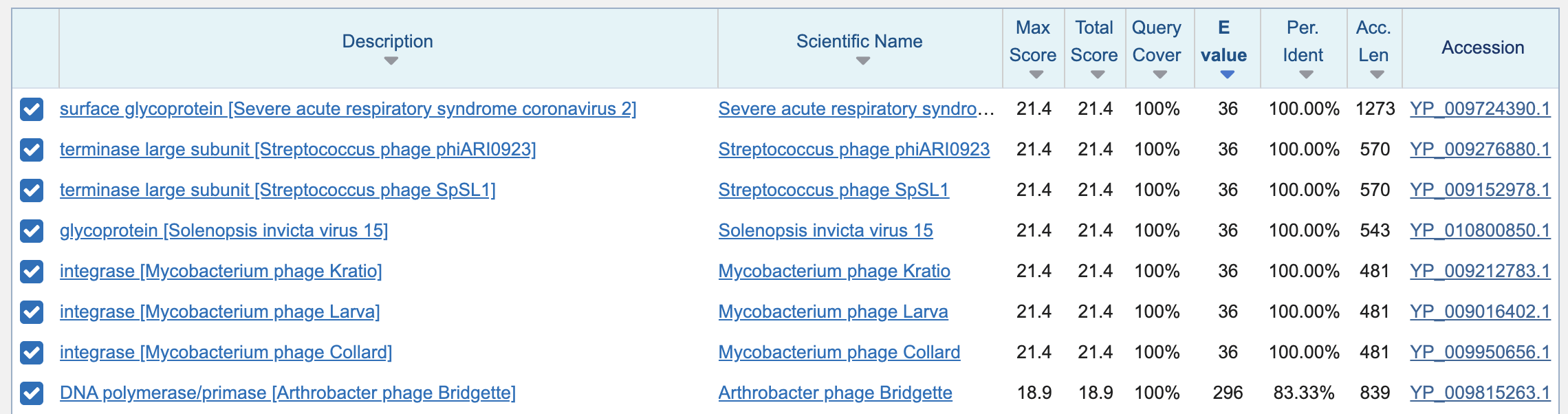
The results for my query above didn't include any HIV sequences,
because the envelope protein of the HIV-1 refseq matches only 2 of 6
residues of TNGTKR at the spot where Pradhan's HIV
sequences from Thailand match TNGTKR.
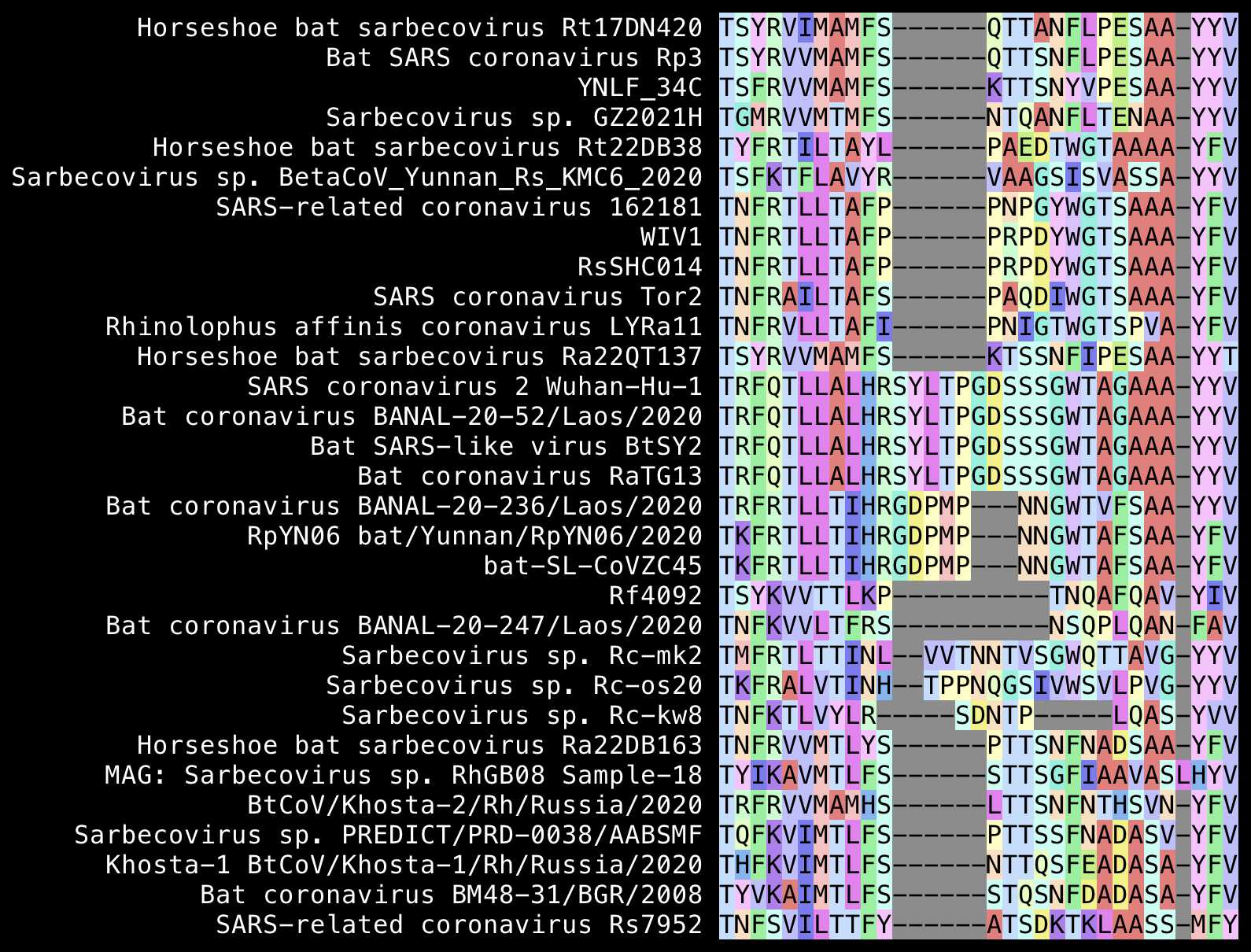
Pradhan identified the locations of the so-called inserts by doing a pairwise alignment of SARS-CoV-2 against SARS-CoV, so his pairwise alignment didn't make it clear if the regions with gaps were inserts in SARS-CoV-2 or deletions in SARS-CoV. He should've done a multiple sequence alignment of a larger set of sarbecoviruses instead, which would've had the additional benefit that it would've inserted the gaps at more accurate positions that would've been informed by the overall phylogeny of sarbecoviruses.
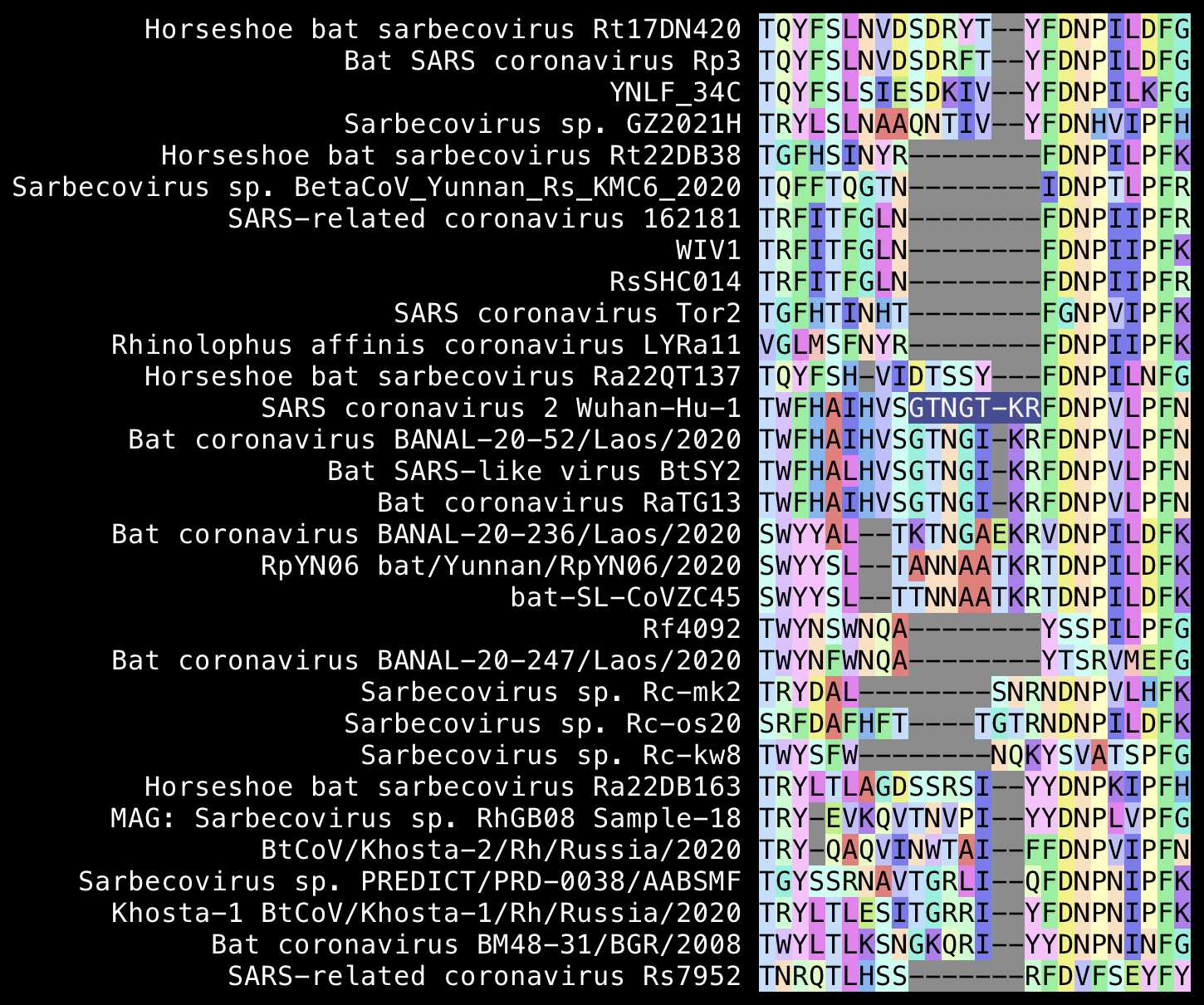
The first so-called insert in Pradhan's alignment was
GTNGTKR, which actually looks more like a deletion in
SARS-CoV than an insertion in SARS-CoV-2, because for example the
Bulgarian virus BM48 has NGKQR at the same spot. The
TKR at the end of the first so-called insert is also
included in ZC45 and RpYN06:
Pradhan's second insert was YYHK, and when he searched
for the insert and surrounding residues on BLAST, he found an HIV
sequence from Kenya that matched the last 2 residues of the insert and
the next 4 residues (HKNNKS).
However the YYHKNNK without the K in the
middle is also included in ZC45 and ZXC21, which had already been
published in January 2020 when Pradhan wrote his paper, so a major
omission in his paper was that he didn't say anything about ZC45 or
ZXC21 in the paper:
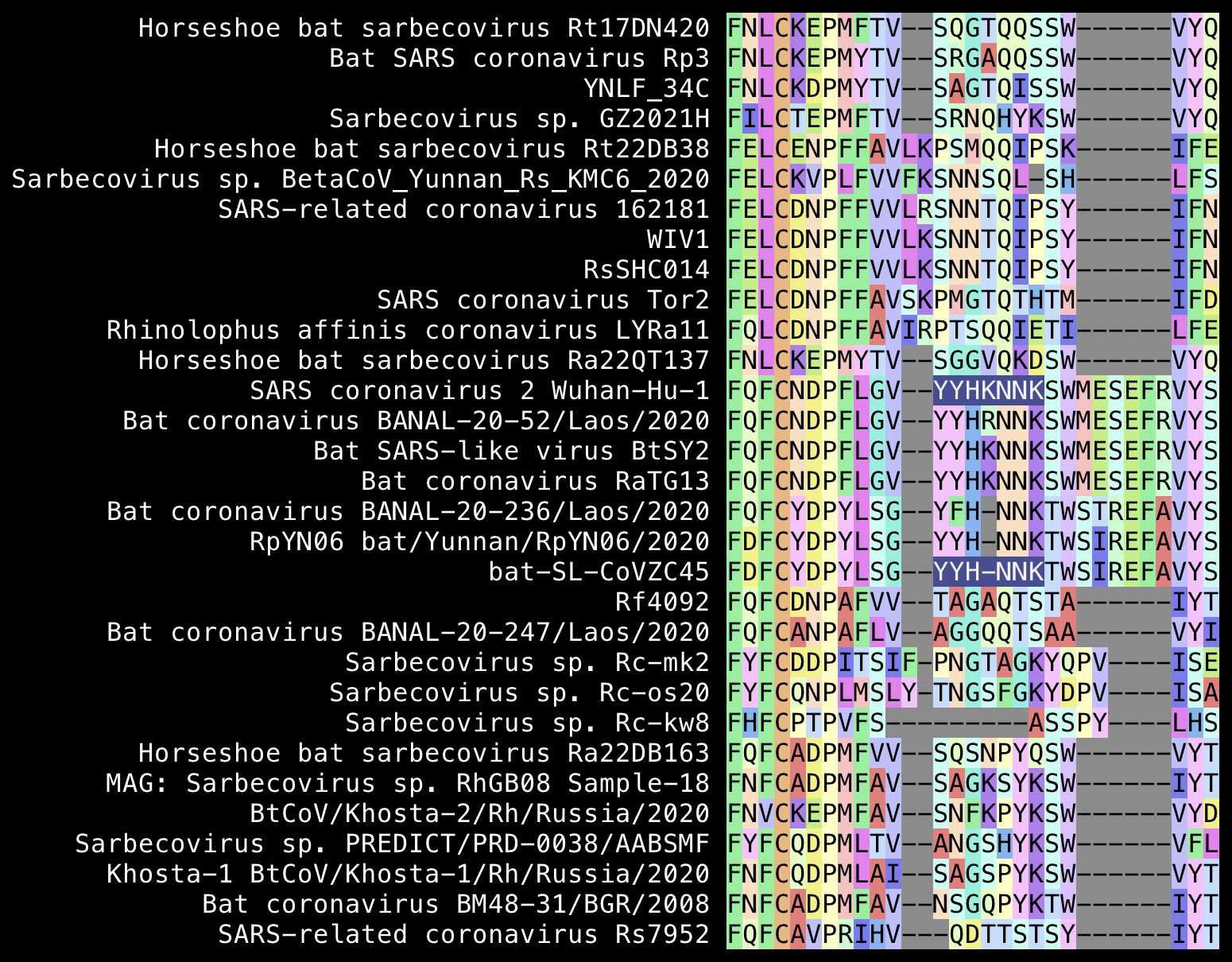
From the alignment above, you can also see that relative to the
original SARS-CoV, SARS-CoV-2 has the deletion SK and the
insertion MESEFR, but the YYHK doesn't even
look like an insert in my alignment. Trevor Bedford also pointed out
that the YYHK didn't look like an insert relative to other
sarbecoviruses, but from his alignment it looks like SARS-CoV-2 has the
insertion SEFR relative to SARS-CoV-: [https://

The third insert Pradhan highlighted in his alignment consisted of
the 3 amino acids SSG, even though a bit before it there
was another insert of 3 amino acids that he ignored in his paper, which
I'm calling insert 2.5. In my alignment below, the gaps were not placed
at the spot of SSG but a few residues earlier, so that
inserts 2.5 and 3 were combined into a single 6 aa insert
SYLTPG:
I used code like this to make the colorized amino acid alignments:
#download whole genome sequences of sarbecoviruses curl -sL sars2.net/ f/ sarbe. fa. xz| xz -dc> sarbe. fa # efetch multiple sequences with accessions from STDIN emu()(curl -sd " id=$( paste -sd,) " " https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=${ 1:- nuccore} & rettype=${ 2:- fasta} & retmode=$ 3") # download spike protein sequences of sarbecoviruses and rename them to have same the defline as the whole genome sequences seqkit seq -ni sarbe.fa| emu ' ' fasta_ cds_ aa| seqkit grep -nrp gene=S, spike, surface| sed ' s/_ prot_.*//; s/ lcl|// '| seqkit fx2tab| awk -F\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ print" > "$ 1" " a[$ 1] "\ n"$ 2} ' <( seqkit seq -n sarbe. fa| sed $' s/ /\ t/ ') -> spike. aa # make a TSV matrix for percentage identity between spike protein sequences aln()(\mafft -- thread 3 -- quiet "${@--} ") curl -Ls sars2. net/ f/ pid. cpp> pid. cpp; g++ pid. cpp -O3 -o pid seqkit replace -isp '[^ X] ' -r ' ' spike. aa| seqkit seq -m 5| seqkit seq -ni| seqkit grep -vf- spike. aa| aln|./ pid> spike. pid # thin out a TSV percentage identity matrix by removing rows with over n% thin()(identity to any previous row (default 99%) awk -F\\ t ' NR> 1{ for( i=2; i< NR; i++) if($ i> x) next; print$ 1} ' x="${ 1- 99} " "${@: 2} ") # print color scheme aacolor()(printf %s\\ n A: 242: 121: 121 C: 242: 182: 121 D: 242: 242: 121 G: 182: 242: 121 F: 121: 242: 151 E: 121: 242: 222 T: 121: 182: 242 I: 121: 121: 242 K: 182: 121: 242 L: 242: 121: 242 M: 255: 191: 191 N: 255: 223: 191 P: 255: 255: 191 Q: 223: 255: 191 R: 191: 255: 207 S: 191: 255: 244 H: 191: 223: 255 V: 191: 191: 255 W: 223: 191: 255 Y: 255: 191: 255 -: 140: 140: 140 X: 255: 255: 255 \*: 255: 255: 255| tr : \ ) # display alignment with colorized amino acids seqcol()(seqkit seq -u| seqkit fx2tab| gawk -F\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ name[ FNR] =$ 1; split($ 2, z, " "); for( i in z) seq[ FNR][ i] =z[ i]; if( length($ 1) > max1) max1=length($ 1); if( length($ 2) > max2) max2=length($ 2)} END{ for( i=1; i< =max2; i+ =width){ printf(