Peter Duesberg said that HIV has been isolated and sequenced but that HIV is a harmless passenger virus which is not the cause of AIDS. I have coined the term "neo-Duesbergians" to refer to the Duesbergian camp of conspiracy theorists who claim that the SARS-CoV-2 virus was not the cause of the deaths that were attributed COVID, even though the neo-Duesbergians either say that the SARS-CoV-2 virus exists, or that it is not important if the virus exists or not, or that they don't know if the virus exists or not. I have also referred to the same group of people as the "no-pandemic" camp since one of their mantras is that there was no COVID pandemic, and I have referred to them as the "no-virus-lite" camp, because they hold many of the same views as the people who claim that viruses do not exist, and because a few luminaries of the camp like J.J. Couey and Mike Yeadon have said that they became enlightened about the lack of a pandemic after they looked into what the no-virus people were saying.
People from the neo-Duesbergian/no-pandemic/no-virus-lite camp have suggested that the deaths attributed to COVID were caused by vaccines, by ventilators, by drugs like Remdesivir or midazolam, by rebranded influenza, by bacterial pneumonia, by reduced prescription of antibiotics, by supplementary oxygen treatment, by psychological stress, by heat waves in the summer, by opioid overdoses, or by some kind of a release of unspecified toxins.
This HTML file consists of my comments to people who claim that there was no pandemic, or that the deaths attributed to COVID were not caused by SARS-CoV-2. I originally published this file because Martin Neil asked me to publish an article about my comments to his Substack.
Further updates are added to part 2: nopandemic2.
A common cutoff for determining whether two viruses belong to the same species or not is whether the whole genome sequences of the viruses have over 90% nucleotide identity.
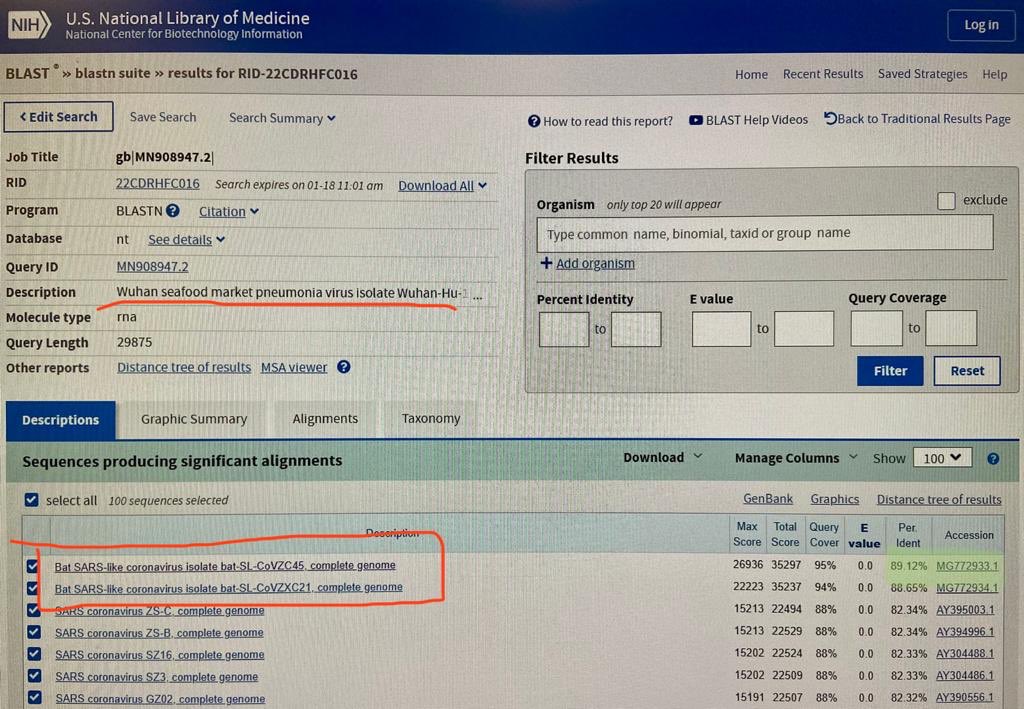
If in January 2020 you did a BLAST search for the genome of SARS-CoV-22, the best match was the bat virus ZC45, which has about 88% identity with Wuhan-Hu-1 if you ignore positions where either sequence has a gap. For example in a tweet on January 18th 2020 UTC, Dinggang Wang posted the following photo of a BLAST search for the genome of SARS-CoV-2: [https://
There is no full genome sequence of a virus which was published at GenBank before 2020 and which has over 90% nucleotide identity with Wuhan-Hu-1 (unless there is some secret genome sequence which had been deleted before early 2020 and which was never discovered by COVID researchers). The RdRp sequence of RaTG13 was published at GenBank in 2016 but it is not a full genome. [https://
The following code downloads sarbecoviruses with a publication date in 2019 or earlier from GenBank, and it then does a multiple sequence alignment of the viruses along with Wuhan-Hu-1, and it calculates the percentage identity of each virus to Wuhan-Hu-1 so that positions where either sequence has a gap are ignored. You can see that ZC45 ranks highest with about 88.11% identity (even though if you do a pairwise alignment of only ZC45 and Wuhan-Hu-1, they get about 88.15% identity):
$brew install seqkit mafft [...] $ curl ftp:// ftp. ncbi. nlm. nih. gov/ entrez/ entrezdirect/ install- edirect. sh| sh [...] $ curl -s ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=fasta& id=MN908947. 3' > sars2. fa $ esearch -db nuccore -query '( viruses[ filter] AND sarbecovirus) 0: 2019[ dp] '| efetch -format fasta> sarbe19. fa $ seqkit seq -m 25000 sarbe19. fa| cat sars2. fa -| mafft -- thread 4 -- reorder -> sarbe19. aln [...] $ pid1()( seqkit fx2tab "$@ "| awk -F\\ t ' NR==1{ split($ 2, a, " "); l=length; next}{ split($ 2, b, " "); d=0; n=0; for( i=1; i< =l; i++) if( a[ i]! ="- " & & b[ i]! ="- "){ n++; if( a[ i]! =b[ i]) d++} print 100*( 1- d/ n),$ 1} ') $ pid1 sarbe19. aln| sort -rn| head 88. 1114 MG772933. 1 Bat SARS- like coronavirus isolate bat- SL- CoVZC45, complete genome 88. 0757 MG772934. 1 Bat SARS- like coronavirus isolate bat- SL- CoVZXC21, complete genome 80. 5101 KF294457. 1 Bat SARS- like coronavirus isolate Longquan- 140 orf1ab polyprotein, spike glycoprotein, envelope protein, membrane protein, and nucleocapsid protein genes, complete cds 80. 1549 AY395002. 1 SARS coronavirus LC5, complete genome 80. 1549 AY395001. 1 SARS coronavirus LC4, complete genome 80. 1549 AY395000. 1 SARS coronavirus LC3, complete genome 80. 1514 AY394999. 1 SARS coronavirus LC2, complete genome 80. 0401 EU371564. 1 SARS coronavirus BJ182- 12, complete genome 79. 979 EU371563. 1 SARS coronavirus BJ182- 8, complete genome 79. 979 EU371561. 1 SARS coronavirus BJ182b, complete genome $ seqkit grep -nrp ' ZC45.* complete' sarbe19. fa| cat sars2. fa -| mafft -- quiet -- thread 4 -| pid1 88. 1459 MG772933. 1 Bat SARS- like coronavirus isolate bat- SL- CoVZC45, complete genome
If SARS-CoV-2 was not a novel virus, then was MERS-CoV not novel either? For about half a year until MERS-CoV was named, it used to be called "novel coronavirus" or "nCoV" or even "novel SARS-like coronavirus": [https://

Other merbecoviruses like HKU4 and HKU5 had been discovered before MERS-CoV, but MERS-CoV has only about 71% identity with HKU4 and HKU5. So the reason why MERS-CoV was called a novel virus was because it actually represented a newly-described species of virus.
If SARS-CoV-2 would have been in widespread circulation long before 2020, then the genomes of SARS-CoV-2 samples collected in early 2020 would be more diverse, except perhaps in the scenario where the earlier undocumented strains would've been replaced by the Wuhan strain, like how Omicron ended up replacing pre-Omicron strains.
But actually there's 54 SARS-CoV-2 sequences that were submitted to GISAID in January 2020, but only 2 samples have more than 10 mutations from Wuhan-Hu-1, and even those are likely to be the result of sequencing or assembly errors:
$curl https:// sars2. net/ f/ gisaid2020. tsv. xz| xz -dc> gisaid2020. tsv $ awk -F\\ t '$ 3~ " 2020- 01" & &$ 10==" Human" ' gisaid2020. tsv| cut -f3- 8, 11- 13| tr \\ t \| 2020- 01- 11| 2019- 12- 24| B| China| Hubei| Wuhan| 3| A3778G, A8388G, T8987A| 2020- 01- 30| 2019- 12- 26| B| China| Hubei| Wuhan| 2| C6968A, T11764A| 2020- 01- 10| 2019- 12- 30| B| China| Hubei| Wuhan| 0|| 2020- 01- 10| 2019- 12- 30| B| China| Hubei| Wuhan| 2| G20670A, G20679A| 2020- 01- 11| 2019- 12- 30| B| China| Hubei| Wuhan| 0|| 2020- 01- 18| 2019- 12- 30| B| China| Hubei| Wuhan| 2| G21316A, A24325G| 2020- 01- 18| 2019- 12- 30| B| China| Hubei| Wuhan| 2| G7016A, A21137G| 2020- 01- 18| 2019- 12- 30| B| China| Hubei| Wuhan| 0|| 2020- 01- 18| 2019- 12- 30| B| China| Hubei| Wuhan| 2| A8001C, C9534T| 2020- 01- 19| 2019- 12- 30| B| China| Hubei| Wuhan| 1| T21656A| 2020- 01- 21| 2019- 12- 30| B| China| Hubei| Wuhan| 0|| 2020- 01- 21| 2019- 12- 30| B| China| Hubei| Wuhan| 1| T6996C| 2020- 01- 21| 2019- 12- 30| B| China| Hubei| Wuhan| 6| T104A, T111C, T112G, C119G, T120C, G124A| 2020- 01- 12| 2019- 12- 31| B| China||| 0|| 2020- 01- 30| 2019- 12- 31| Unassigned| China| Wuhan|| 25| C344T, T445A, G1167C, G2408T, C2881A, G4127T, A4426C, T6000A, C6593T, A6948G, C8320T, T10061C, T10062G, G10610T, G12311T, C12318G, G12332T, G12338C, G12345T, T12346G, G13445C, A14052T, T14073A, C23730T, T25535A| 20618- 20622, 26170- 26182 2020- 01- 11| 2020- 01- 01| B| China| Hubei| Wuhan| 2| C27493T, C28253T|