See these pages:
Fleming wrote: "I received my PhD two days after
my HS diploma. That was the requirement from the work begun in 1968 when
our group, following the MK Ultra group, was initiated. I owe much to
many including respect and confidentiality of our work." [https://
When someone asked Fleming to post his PhD thesis to show it wasn't
fake, Fleming replied he couldn't show the thesis because it was
classified under a classification level requiring Q-clearance. [https://
The Twitter user LogarithmicDis found many problems with Fleming's
diploma: [https://
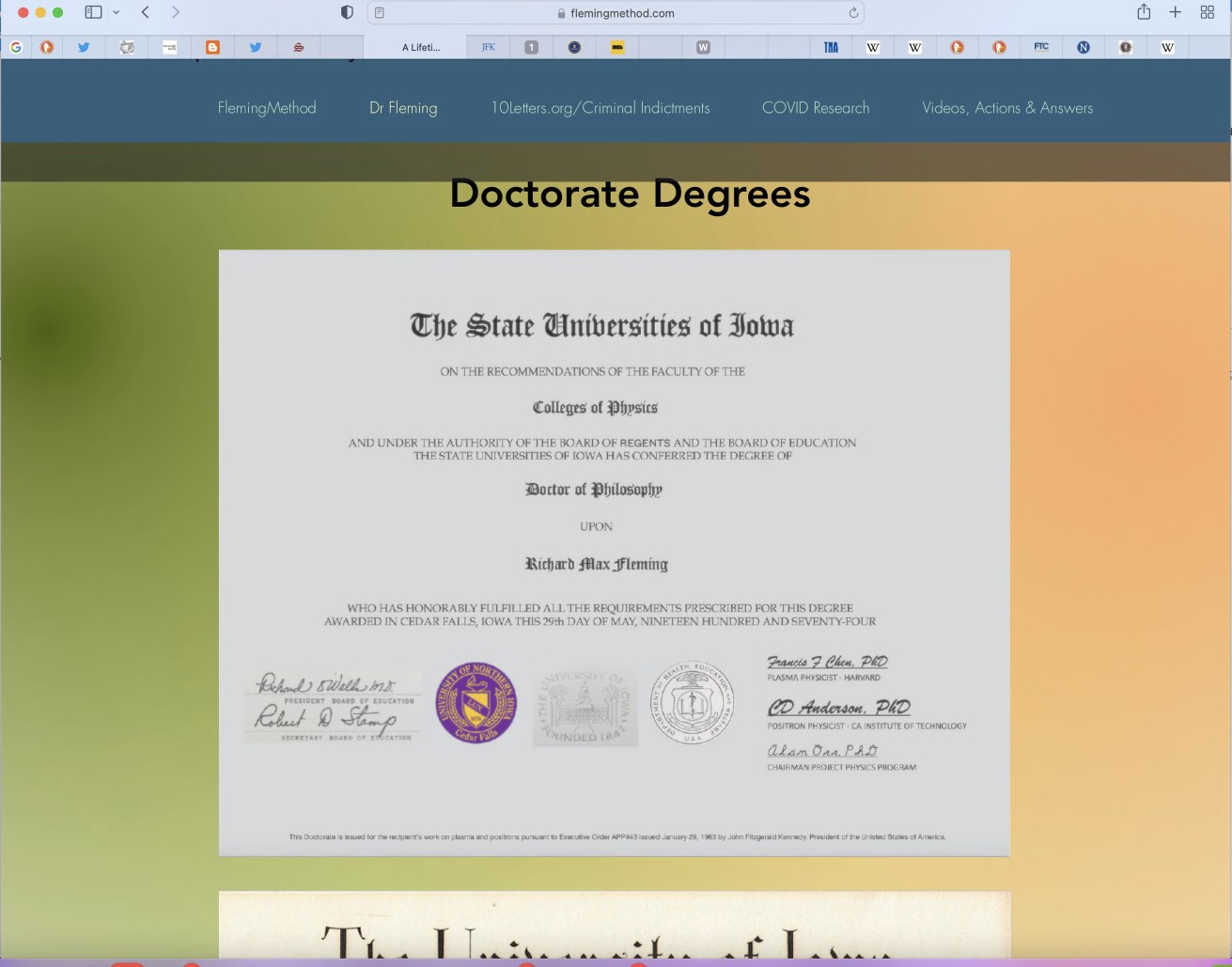
On his website he has posted his "highly unusual" PhD diploma that he started on supposedly at age 12, if you subtract his birth year (posted online by him) from a 1968 matriculating date = 12 years old.
But the diploma has MANY problems.
First, the doctor is a convicted felon and was sentenced for his criminal acts and debarred by the FDA.
https://
archives. fbi. gov/ archives/ omaha/ press- releases/ 2009/ om082009. htm 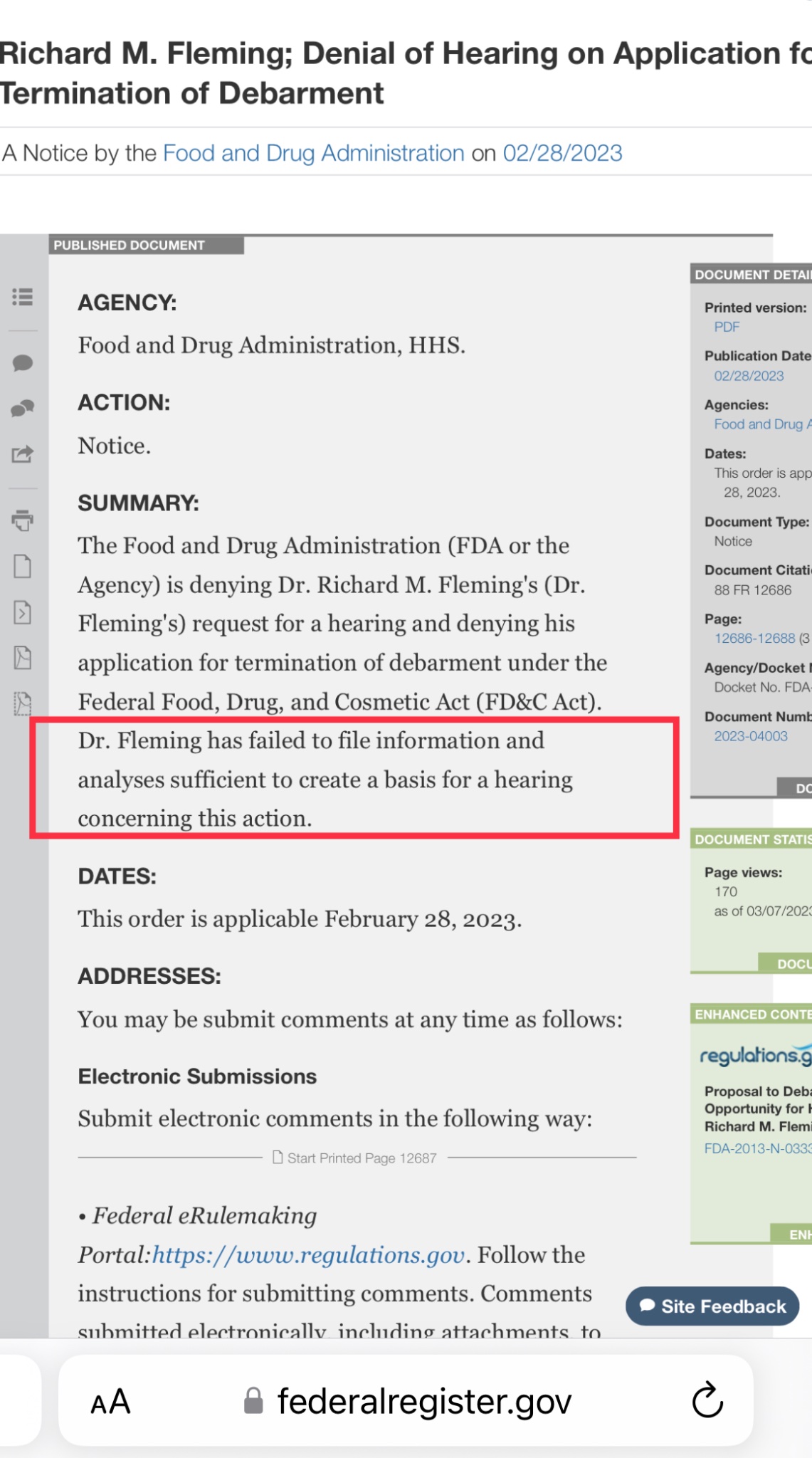
So his honesty isn't exactly batting a thousand here. Upon closer look at his CV posted on his website:

And closer look at the unusual PhD diploma with contrast adjusted, we see a copy and paste left hand signature rectangle and very unusual seals.
But there are 12 total problems with this "diploma" that I see:

1. There is no such thing as "The State Universities of Iowa".
University of Iowa was in fact called "State University of Iowa".
Singular, not plural.
https://
iowaregents. edu/ the- board/ regents- history 
2. The copy & paste left hand signatures (contrast adj).
3. The president of the Board of Regents was Mary Peterson in 1974. So wrong name here.
4. The proper title is President, Board of Regents (not Board of Education, a term that ceased in 1955).
https://
iowaregents. edu/ the- board/ regents- history 
5. The seals appear to not be original to the background
6. LT and RT seals do not fade w/the document (along w/the square patch on left and e-signatures in right, addressed later).
7. The middle seal appears curved and uneven, like a poor patch job.
8. Right seal is what?
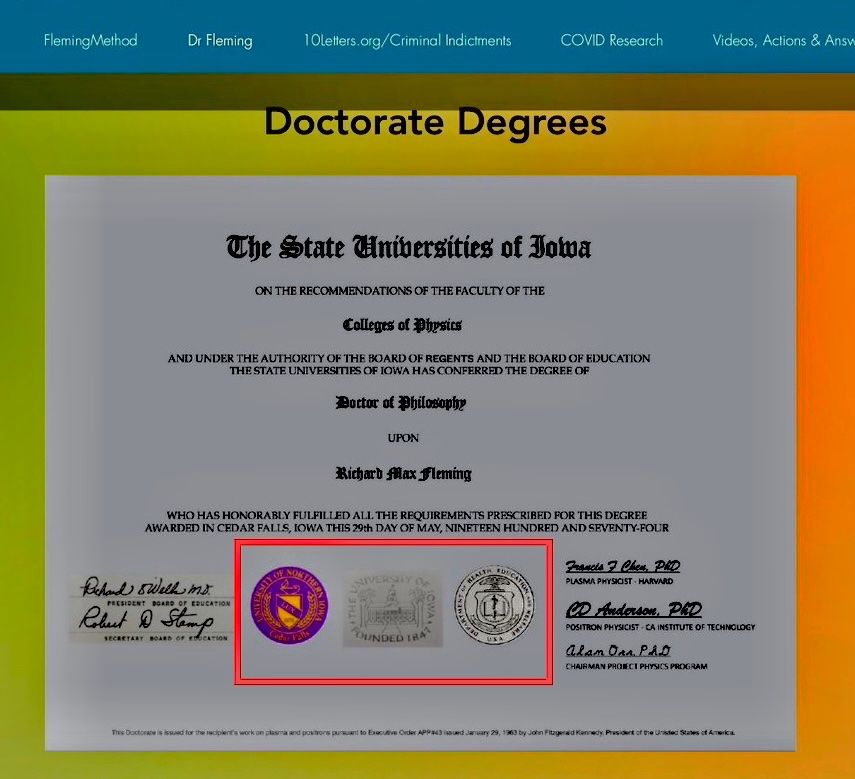
9. Two electronic signatures are exactly the same font and uncharacteristic of 1974.
10. Weird fine print at bottom with misspelling.
11. Executive order reference to JFK is strange on a diploma. Here are JFK's EOs:
https://
en. wikipedia. org/ wiki/ List_ of_ executive_ actions_ by_ John_ F% 2e_ Kennedy 
12. UNI does not confer PhD degrees. It was formerly a teachers college and confers only EdD degrees.

Here's his CV from @Doctor_I_am_The federal criminal court case in 2009.
He failed to update it.
Certification Board in Nuclear Cardiology reveals no certifications currently.
https://
apca. org/ certifications- examinations/ cbnc- and- cbcct/ cbnc/ ASNC says expired in 2002.

I verified the diploma was fake with the regent universities. I actually called (like that was necessary? lol 😂)
[...]
And the ridiculous statement at the bottom of Fleming's confirmed fake degree is a complete joke.
Here are JFK's EO's that day.
https://
federalregister. gov/ presidential- documents/ executive- orders/ john- f- kennedy/ 1963 
The line at the bottom of the diploma says: "This Doctorate is issued for the recipient's work on plasma and positrons pursuant to Executive Order APP#43 issued January 29, 1963 by John Fitzgerald Kennedy, President of the Unisted [sic] States of America." But the list of executive orders issued by JFK is public, and it doesn't include any order number "APP#43".
I tried to check if the diploma featured fonts which had not yet been published in 1974, but I believe the two fonts I was able to identify had both been published before 1974 (even though it's possible that Fleming would have used newer near-identical variants of the old fonts I identified, because there's newer copycat versions of both fonts.)
The signatures of Francis F Chen and CD Anderson seem to have been written in some version of the Brush Script font, which was designed in 1942:
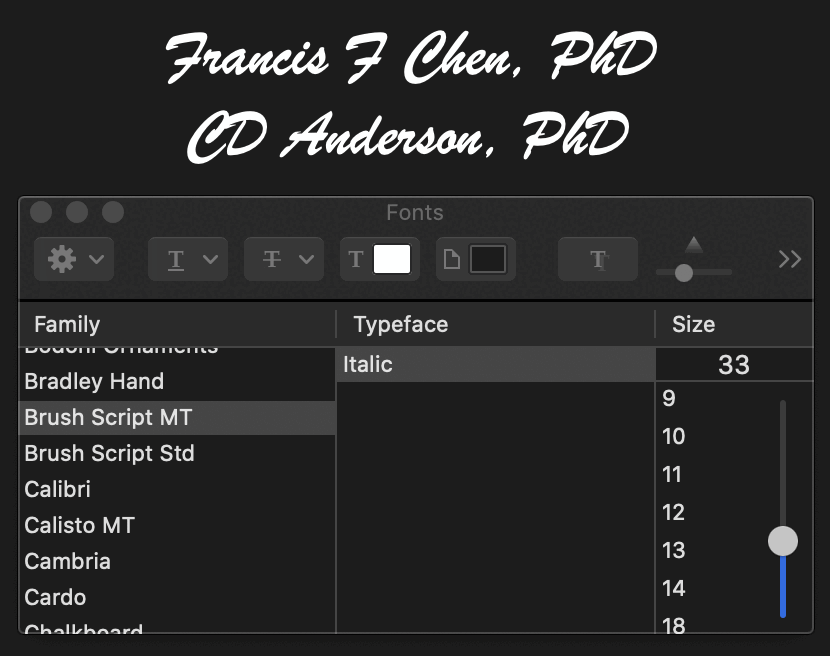
At first I thought the blackletter font in the diploma was Monotype
Old English Text which was only published in 1990. [https://

When I linked to the thread by LogarithmicDis, Fleming told me: "This image of my doctoral diploma is clearly altered and
you are either guilty of altering it or guilty of presenting an altered
diploma. It's a new country. :-)". [https://
But perhaps Fleming was projecting his own sins onto his adversaries,
because at some point after LogarithmicDis posted her thread, Fleming
modified the image of his diploma on his website, so that he altered the
background of the signatures on the left and the two seals in the middle
to match the background of the rest of the paper: [https://

The old version of the image where the backgrounds don't match is
still available at the Wayback Machine: [http://

When I asked Fleming about his PhD thesis, he told me to read his
book "Are We the Next Endangered Species".
[https://
Fleming used to have a Twitter account called ExperiencedStud, where
he advertised his work as an actor and he tried to get casted in adult
films. There's no snapshots of the account at the Wayback Machine, but
Cheshire managed to take these screenshots of the account: [https://
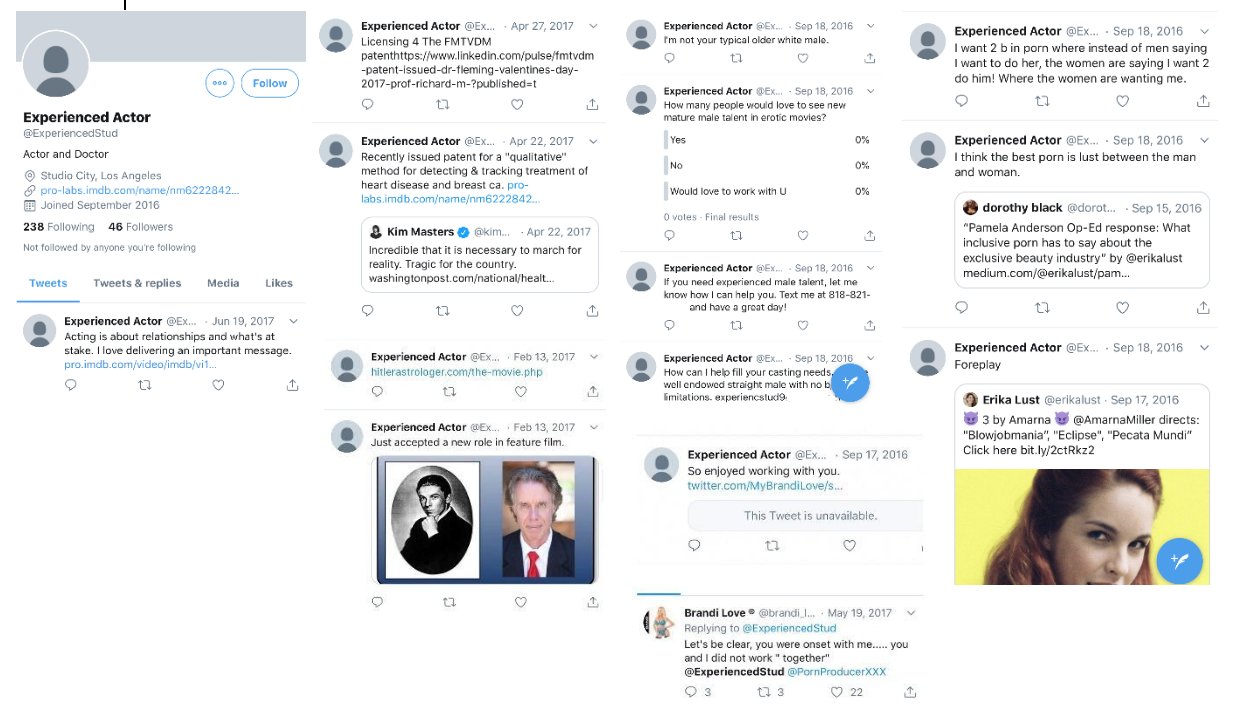
I don't know if Fleming actually managed to appear in any adult
movie, because one of his tweets from 2016 said: "I've been acting a couple years. Just started in porn a
few weeks ago after people told me for years I'm a natural. Looking to
be your talent". [https://
Fleming said that the account was hacked, and that when he was called
an adult actor, it just meant that he was not a child actor: [https://
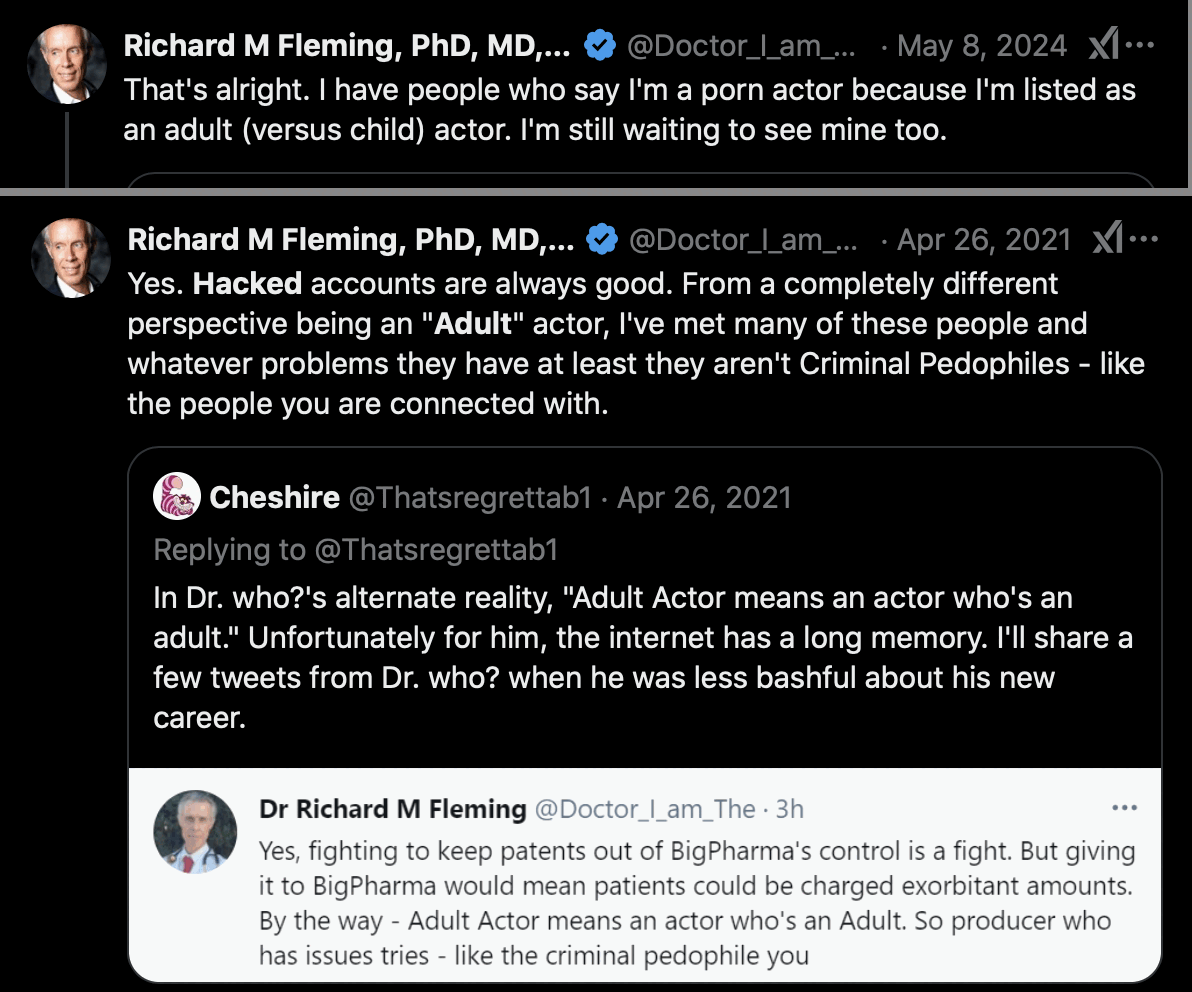
Fleming also claimed that the account was called "Experienced Student that got truncated and then
hacked": [https://
The truncation theory is actually somewhat plausible, because when you register a new account on Twitter, you are first asked to enter a display name, and then Twitter automatically suggests a username based on the display name, so that spaces are removed and the name is truncated to at most 15 characters, and "ExperiencedStud" happens to be 15 characters long. But why would Fleming not have edited the username manually?
However as evidence against Fleming's hacking theory, the court
documents of the Fleming v. Sims case said: "According to Larry Sims, the parties first came into
contact after Defendants placed a job posting on an adult entertainment
job listing website. (See, e.g., ECF Nos. 90 at 1-2,
124 at 1, 188 at 1, 234 at 1, 244 at 1.) Plaintiff allegedly
responded to the advertisement and represented himself as an adult film
actor and model. (Id.)" [https://
As an aside, the reason why Fleming said that Cheshire was connected
with "criminal pedophiles" was because David
Gorski's blog used to be hosted on a platform called ScienceBlogs, which
was operated by a company called Seed Media Group that published a
science magazine called Seed. Epstein had invested in Seed Media Group,
and Ghislaine Maxwell was briefly on their board of directors in
2007-2008. But Gorski wrote that he didn't know at the time that Epstein
or Ghislaine were connected to the blogging platform (and at the time they were not yet household names, so
it's likely that Gorski wouldn't have even know who they were).
[https://
Fleming's Twitter account used to be called MedicinesDarkK1 until
October 2020. [https://
Fleming's IMDB profile has a photo where he poses as Batman and a
video where he poses as Joker: [https://
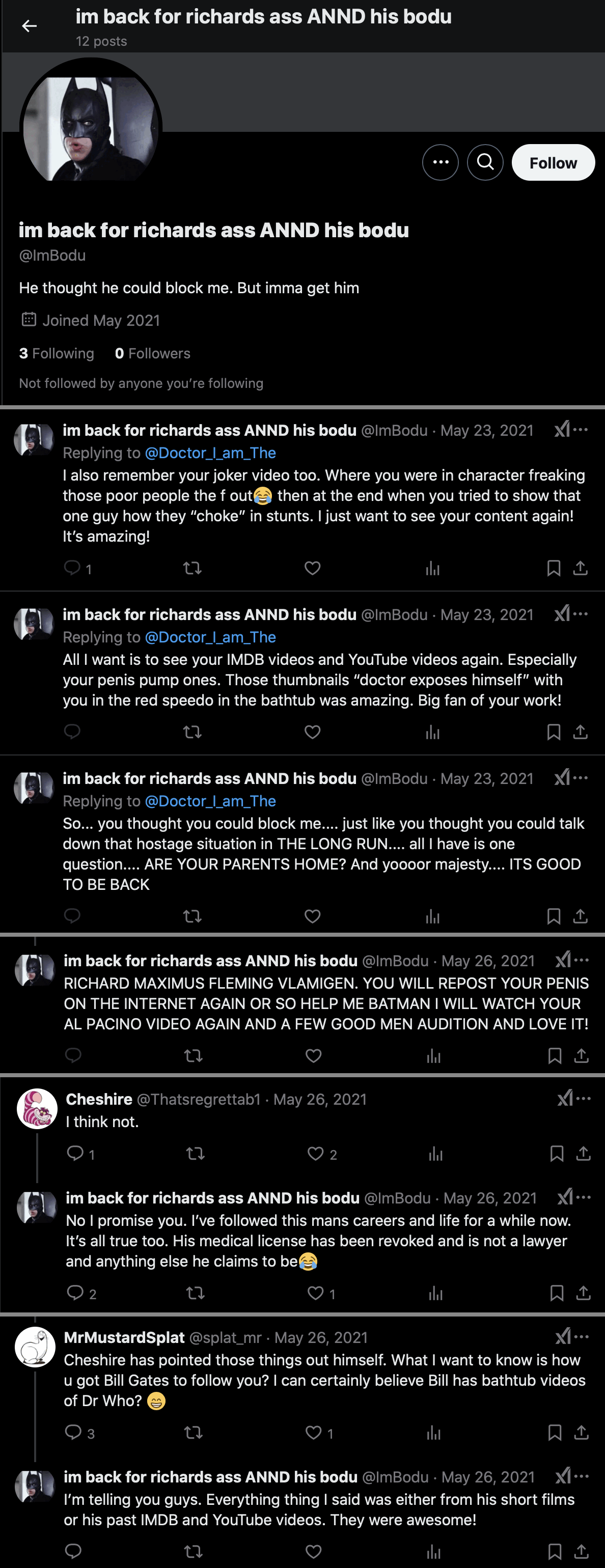
There's an account with a Batman avatar called "ImBodu" (where the "Im" seems to match "I_Am" in the username of Fleming's main
account). The ImBodu account was created in May 2021, and it
currently has only 12 tweets. Its display name is "im back for richards ass ANND his bodu" and its
bio says "He thought he could block me. But imma get
him". Cheshire suspected the account might have been created by
Fleming himself. [https://
In 2019 Fleming published a paper titled "Statistical Demonstration that FMTVDM is Superior to
Mammography". [https://
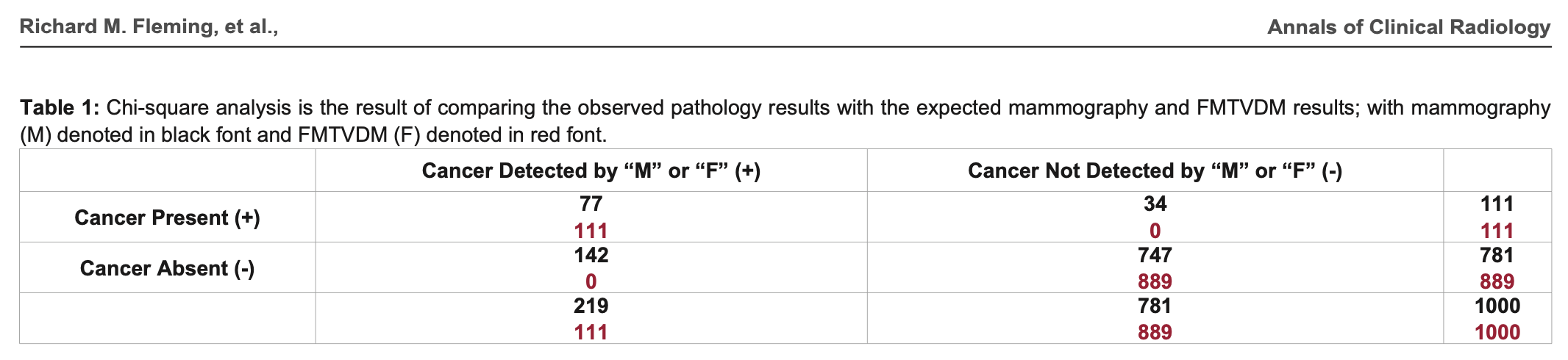
In the table above, the sum on the row for mammography with cancer absent is wrong, because 142 plus 747 should add up to 889 and not 781. But that's the least of Fleming's problems. The formula he used to calculate the chi-squared test is also wrong, and it has two divisions by zero:

In his formula χ² = E (O-, the first E
should've been ∑ and the second number 2 should've been
superscripted: χ² = ∑((.
Fleming also misunderstood the meaning of the expected counts E in the formula. He treated the results of FMTVDM as the expected counts and he compared them to the results of mammography which he treated as the observed counts. But his observed counts should've consisted of a contingency matrix with one row for mammography and another row for his Fleming method. And then he should've calculated the expected matrix based on the observed matrix by doing a matrix multiplication of the row sums and column sums and then dividing the result with the sum of all values in the contingency matrix:
>m=rbind( mammography=c( 77, 34, 747, 142), Fleming_ FMTVDM=c( 111, 0, 889, 0)) > colnames( m) =c( " present_ correct", " present_ incorrect", " absent_ correct", " absent_ incorrect") > m present_ correct present_ incorrect absent_ correct absent_ incorrect mammography 77 34 747 142 Fleming_ FMTVDM 111 0 889 0 > expected=rowSums( m)%*% t( colSums( m))/ sum( m) > expected present_ correct present_ incorrect absent_ correct absent_ incorrect [ 1,] 94 17 818 71 [ 2,] 94 17 818 71
And only after that he should've calculated
sum((:
>sum(( m- expected)^ 2/ expected) 194. 4741 > chisq. test( m, correct=F) # same result Pearson's Chi- squared test data: m X- squared = 194. 47, df = 3, p- value < 2. 2e- 16
Here the procedure of calculating the expected matrix is written out using basic arithmetic operations:
>rowsums=c( 77+ 34+ 747+ 142, 111+ 0+ 889+ 0) > colsums=c( 77+ 111, 34+ 0, 747+ 889, 142+ 0) > sum=c( 77+ 111+ 34+ 0+ 747+ 889+ 142+ 0) > rowsums [ 1] 1000 1000 > colsums [ 1] 188 34 1636 142 > sum [ 1] 2000 > mult=matrix( c( 1000* 188, 1000* 34, 1000* 1636, 1000* 142, 1000* 188, 1000* 34, 1000* 1636, 1000* 142), byrow=T, ncol=4) > mult [, 1] [, 2] [, 3] [, 4] [ 1,] 188000 34000 1636000 142000 [ 2,] 188000 34000 1636000 142000 > mult/ sum # expected matrix [,1] [, 2] [, 3] [, 4] [ 1,] 94 17 818 71 [ 2,] 94 17 818 71
I don't know if it would've been better to just use a simple 2 by 2 contingency matrix with one column for the number of correct detections and another column for the number of incorrect detections, but it gave me close to the same chi-squared value as the test with the 4 by 2 matrix:
>m=rbind( mammography=c( 77+ 747, 34+ 142), Fleming_ FMTVDM=c( 111+ 889, 0+ 0)) > colnames( m) =c( " correct", " incorrect") > m correct incorrect mammography 824 176 Fleming_ FMTVDM 1000 0 > chisq. test( m, correct=F) } Pearson' s Chi- squared test data: m X- squared = 192. 98, df = 1, p- value < 2. 2e- 16 > expected=rowSums( m)%*% t( colSums( m))/ sum( m) > sum(( m- expected)^ 2/ expected) [ 1] 192. 9825
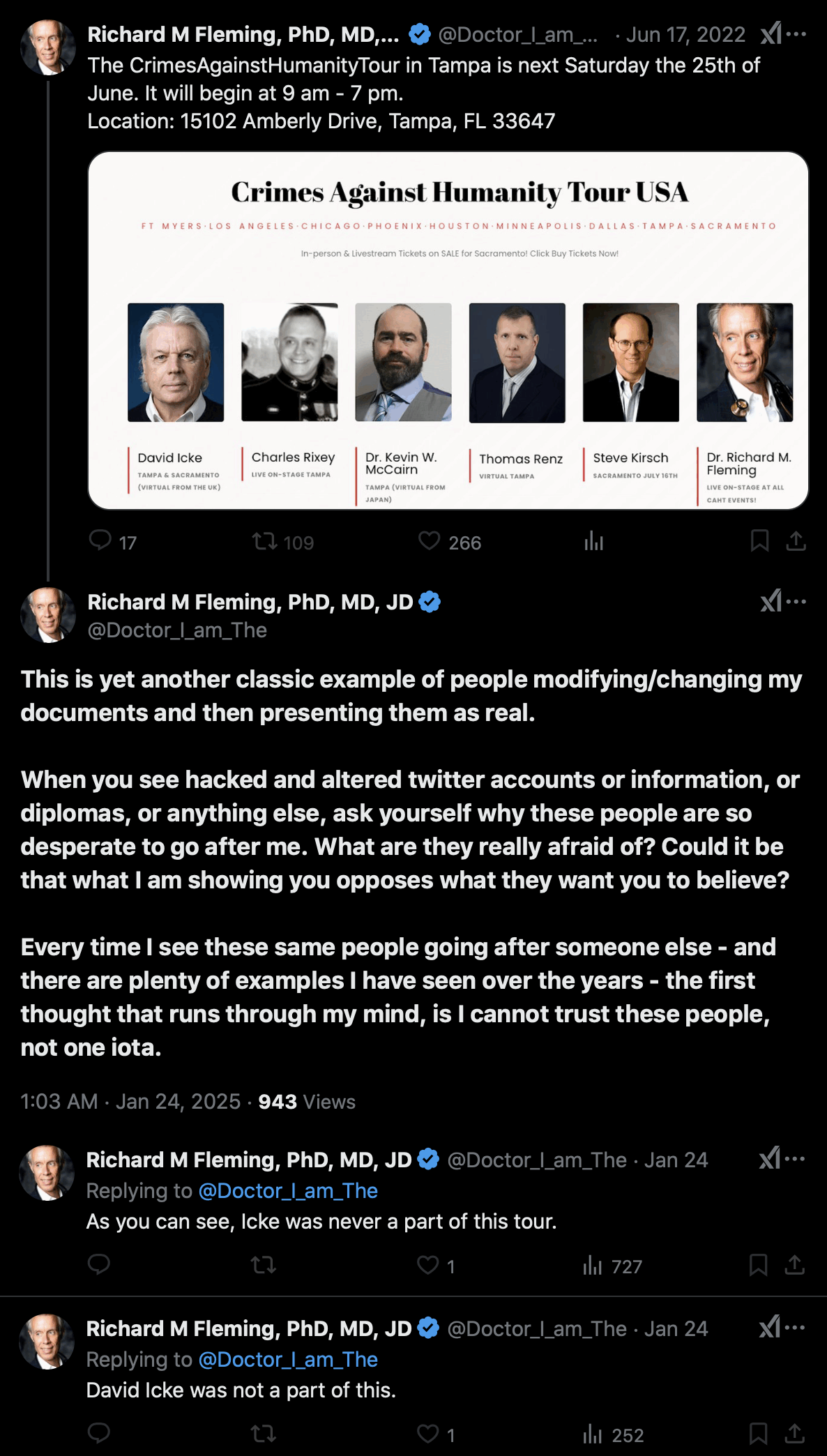
Fleming claimed that his Twitter account had been hacked so that
someone had added a photo of David Icke to a poster for Fleming's Crimes
Against Humanity Tour: [https://
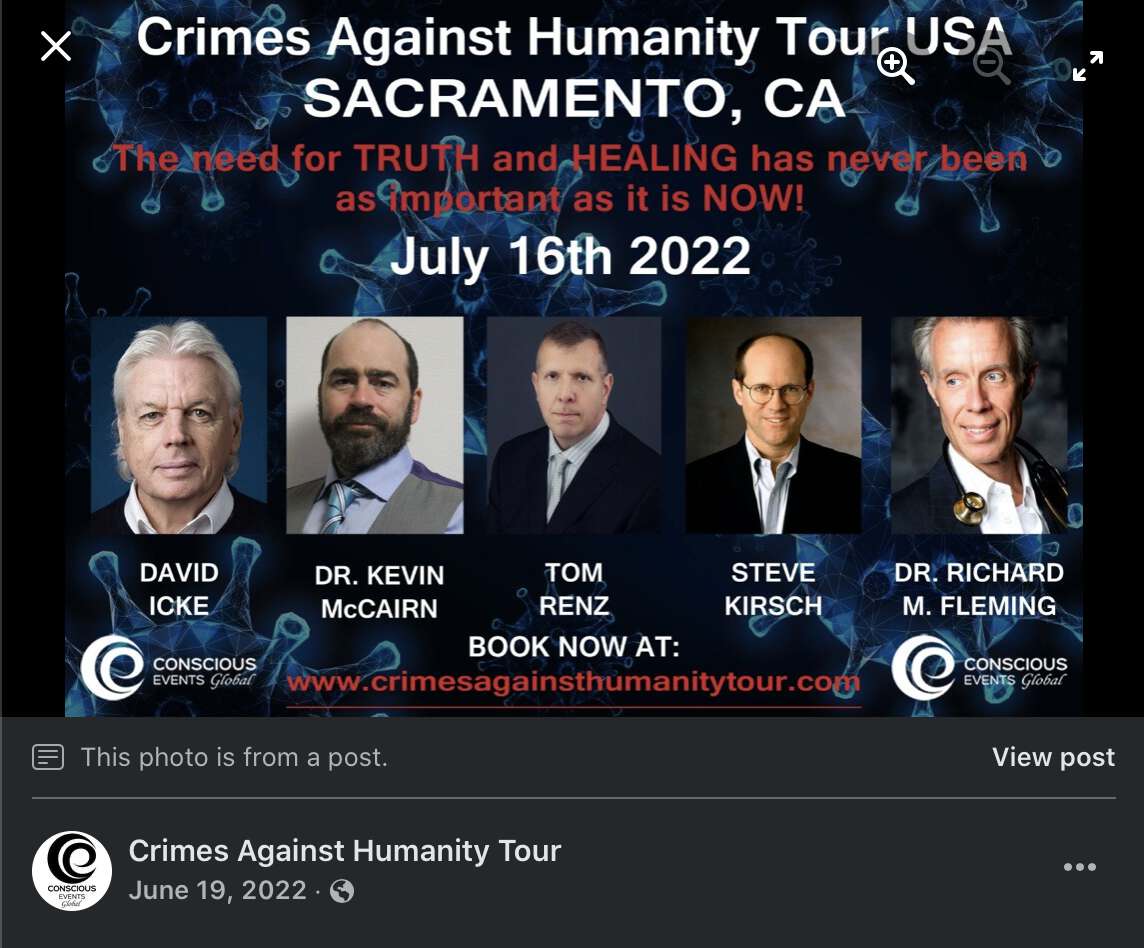
But then was Fleming's Facebook account also hacked? The Facebook
account of the tour also has a poster that features David Icke: [https://
David Icke's blog also said: "I am currently
traveling the country with Drs. Judy Mikovits, Richard Fleming and
Reiner Fuellmich. Our one-day conference topic in nine cities focuses on
the case for crimes against humanity having been committed by leaders of
Big Pharma and the biosecurity cartel." [https://
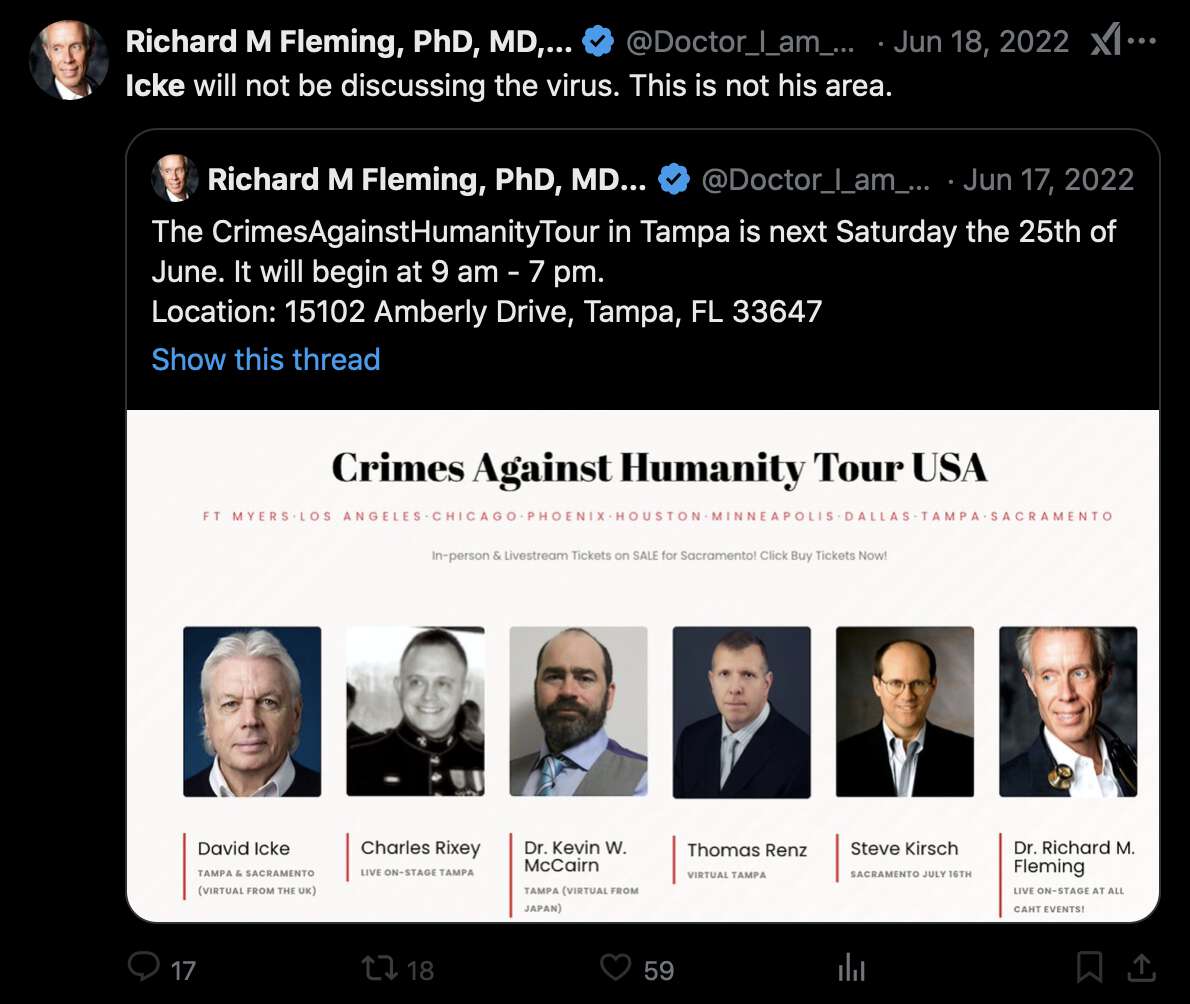
In 2022 Fleming quoted a tweet that featured the poster and referred
to Icke in his tweet. So if the image file of the poster was
photoshopped by the hackers, then did the hackers also edit the text of
Fleming's tweet so that it referred to the photoshopped poster? [https://
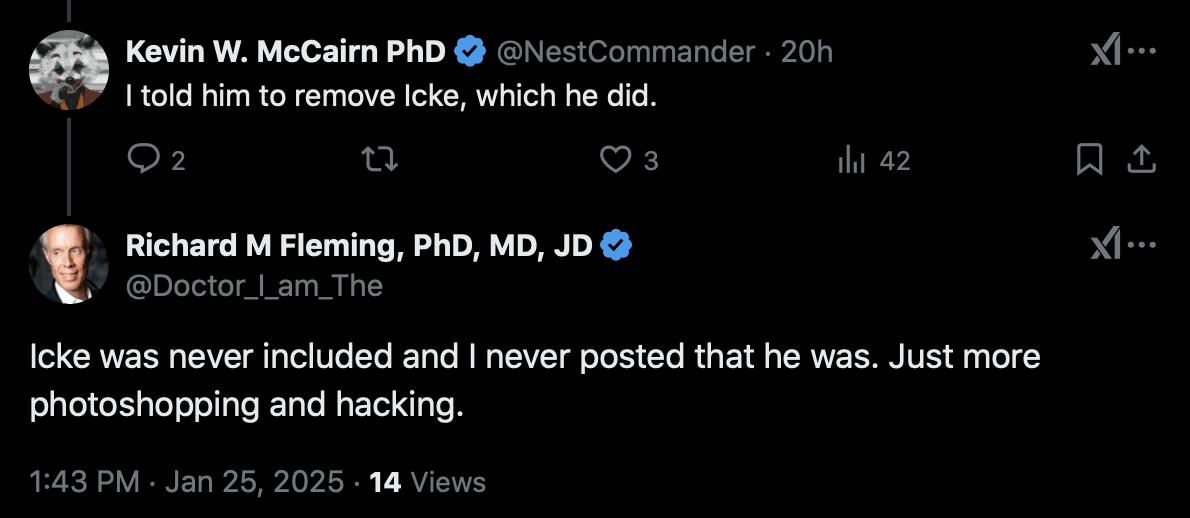
One of the people featured on the tour was Kevin McCairn, who told me
that he asked Fleming to remove Icke from the tour. But even after that,
Fleming still denied that Icke was ever included on the tour, and
Fleming said "more photoshopping and
hacking": [https://
In 2026 Fleming posted a Word document of his supposed PhD thesis on
a preprint server. [https://
Cheshire pointed out that Fleming wrote "Although
the first positron emission tomography (PET)
scanners were built by Edward Hoffman, Michael M. Ter-Pogossian and
Michael E. Phelps last year (1973) at
Washington University with DOE and NIH monies", even though the
DOE wasn't formed until 1977. [https://
Cheshire also found that Fleming cited this textbook: "Goldston, R. J., & Rutherford, P. H. (1969). Plasma Physics. Plasma Physics and
Controlled Fusion, 11(5), 417-435."
Goldston and Rutherford wrote a book titled "Introduction to Plasma Physics", but it wasn't
published until 1995. [https://
Fleming also cited a book with a 13-digit ISBN number, even though
13-digit ISBN numbers were only adopted in 2007. Fleming gave the book
the ISBN-13 number "978-0486449820", which
otherwise matched a 2006 reprint edition of the book, but the check
digit at the end should've been 1 and not 0. [https://
Fleming's thesis committee supposedly consisted of the famous plasma
physicist Francis Chen, the Nobel laureate Carl David Anderson who
discovered the positron, and the biologist Alan Orr who worked at the
University of Northern Iowa, and Fleming's thesis advisor was Alan Orr.
However LogarithmicDis pointed out that Alan Orr was a plant biologist
publishing on maize, and not a physicist. [https://
Before Fleming published the paper about his fake trial in a predatory journal, he published two preprint versions of the paper as well as two accompanying short reports:
April 14th 2020: Fleming submitted a trial to ClinicalTrials.gov
with the title "The Fleming [FMTVDM] Directed CoVid-19 Treatment Protocol (FMTVDM)". [https://
October 20th 2020: Fleming published a preprint about the trial
titled "Is there a treatment for
SARS-CoV-2?". [https://
October 30th 2020: Fleming posted two short reports about his
study as preprints. He described the results of two supposed treatment
arms in a preprint titled "Preliminary Results of
Tocilizumab and Interferon a- 2b Treatment of SARS-CoV-2". [https://
November 12th 2020: Fleming posted an updated preprint about the
trial under the title "Is there a treatment for
SARS-CoV-2? Quantitative Nuclear Imaging finds Treatments for
SARS-CoV-2." [https://
February 18th 2021: The paper was published in a predatory
journal with the title "FMTVDM Quantitative Nuclear
Imaging finds Three Treatments for SARS-CoV-2". [https://
Cheshire posted this comment about the paper at PubPeer: [https://
Highlights: 23 sites, 7 countries, 1,800 patients, 10 treatment arms. No other authors other than Fleming and son. None of the sites were identified, although the ill-formatted tables (apparently due to unfamiliarity with Acrobat) include Table 3 showing the country of the sites. No funding was required for this study (?).
Interestingly, the Clinicaltrials.gov site shows that on October 8, the first author/sponsor changed the "treatment arms and interventions" details on all of the tested protocols. This is 3 weeks after the trial was marked concluded. An example of the changes:
Experimental: Treatment 1 ORIGINAL
Hydroxychloroquine 200 mg po q 8 hrs (600 mg qD) for a total of 10-days, and Azithromycin 500 mg IV on day 1, followed by 250 mg IV on days 2-5 (to prevent bacterial superinfection).
Experimental: Treatment 1 REVISED
Hydroxychloroquine 200 mg po q 8 hrs (600 mg qD) for a total of 10-days , OR Hydroxychloroquine 155 mg IV every 8-hours (600 mg qD) for 10-days if patient is intubated and Azithromycin 500 mg IV on day 1, followed by 250 mg IV on days 2-5 (to prevent bacterial superinfection). (emphasis added)
Source: https://
clinicaltrials. gov/ ct2/ history/ NCT04349410? A=6& B=7& C=merged# StudyPageTop Other remarks:
- While there is mention of centralized institutional board review there is no mention of informed consent by patients or authorized representatives
- There does not seem to be any data provided as to the racial demographics of the patients
- Although 8 different Figures are referenced in the text, none appear in the preprint
- The author acknowledges 6 individuals (by initials only) for their assistance with the "individual patient centers." In comparison, a NEJM-published study on Remdesivir for COVID-19 with 35% fewer patients than this one, acknowledged over 200 physicians by name. https://
www. nejm. org/ doi/ full/ 10. 1056/ NEJMoa2007764
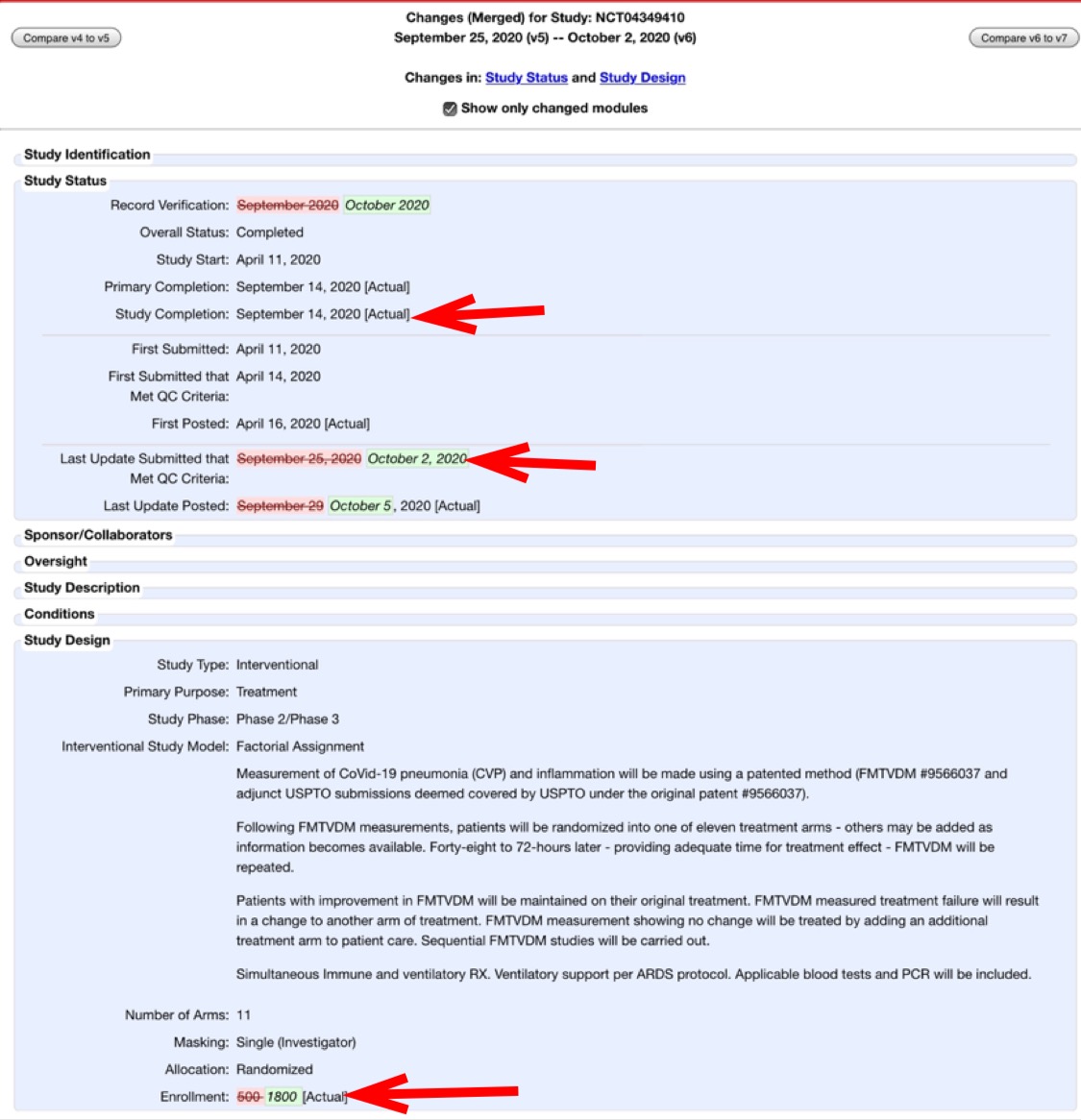
Cheshire also pointed out that after the trial is supposed to have already ended, Fleming changed the number of enrollees from 500 to 501 to 1,800:
Trial ended on September 14; at which time clinical trials.gov showed 500 enrollees.
Dr. Fleming posted this on Twitter on September 24, showing 501 enrollees:
On October 2, the sponsor (Dr. Fleming) changed the trial enrollment number to 1,800 at clinicaltrials.gov and 1,800 is the figure used in this preprint.
However the discrepancy seems to be because Table 3 shows that the total number of patients was 1,800, but 1,299 of them were outpatients and the number of inpatients enrolled to phases I and II was 501:
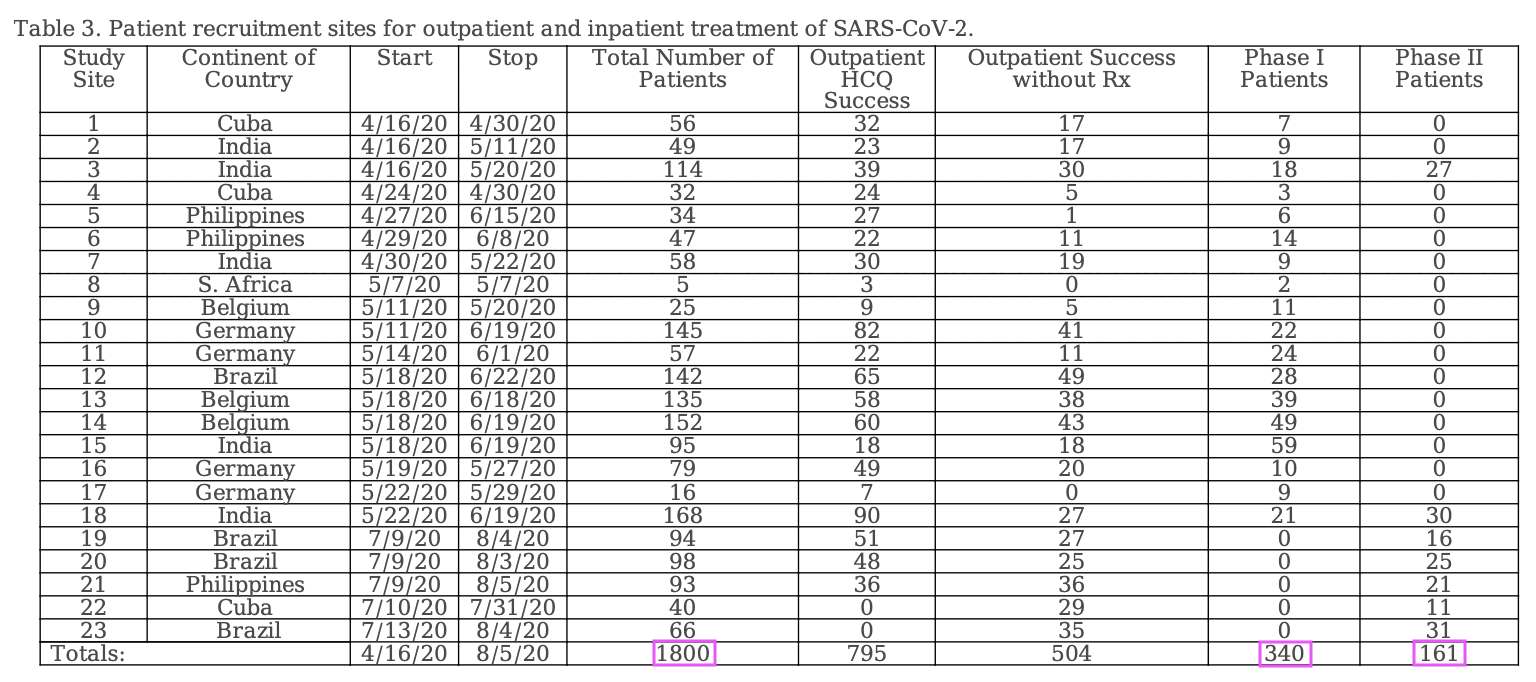
Fleming wrote that "NCT04349410 required identification of a seven-person SARS-CoV-2 treatment team" (where presumably each site would've had its own "treatment team" with at least seven persons, but the study is supposed to have included 23 sites, so 23 times 7 would be 161). Cheshire wrote:
Given the extraordinary requirement that a 7-person treatment team (see below) be formed to treat the 501 hospitalized patients, how is it possible that all this additional care was provided with no funding from the sponsor? Why was this "required identification" not included as part of the pre-registered trial protocols? What evidence does the sponsor have that this "required identification" was followed in every case?

Someone also posted this comment at PubPeer:
It is noteworthy that these ambiguities about the study's feasibility and protocols are reminiscent of earlier skepticism, expressed 18 years ago, about another purported clinical trial from the same author:
...clinical trials are expensive, difficult and time-consuming. Even small dietary trials can easily cost several hundred thousand dollars and require entire research teams. The DPP estimated a cost of $1,075 just to recruit each participant. Fleming reports on a one-year trial of 100 participants and four diets with extensive follow-up. His paper, however, has no co-authors; it acknowledges no source of funding, nor any nurses, dietitians or technicians who might have helped. Fleming identifies himself as Medical Director of Preventive Cardiology, the Camelot Foundation at the Fleming Heart & Health Institute, but if his Web site or receptionist are any indication, he is the sole member of each of those.
As for the issue of peer-review, Fleming states that his patients "were randomly assigned to one of the four dietary regimens based upon dietary preferences." This protocol is pivotal to interpretation of the findings, yet oxymoronic: If patients were assigned to diets based on their dietary preferences, then they weren't randomly assigned. If they were randomly assigned, then their preferences must be irrelevant. The two methods are incompatible. If this paper was peer-reviewed, it was done poorly. If this constitutes high-quality research in this field, then I suggest even more skepticism is necessary.
source: https://
www. [Not behind a paywall here: https://washingtonpost. com/ archive/ lifestyle/ wellness/ 2002/ 09/ 24/ interactions/ dfcd1470- eedb- 474f- 8ae8- 714836e810d9/ www. ]askbjoernhansen. com/ 2002/ 09/ 02/ maybe_ it_ wasnt_ a_ big_ fat_ lie. html
Cheshire wrote:
This trial does not seem to have been registered in India as appears to be required:
My trial is already registered in another Primary Register, then why do I need to register again with the CTRI? A clinical trial being conducted in India, is also required to be registered in the CTRI as the CTRI captures data specific for the Indian arm of a trial, e.g. Site and PI details, Name of Ethics Committee and approval status, target sample size in India, start date in India etc.
Search here: http://
ctri. nic. in/ Clinicaltrials/ advancesearchmain. php
Cheshire wrote: [https://
I'm confused again.
From the Methods section of this preprint (posted 10/27/20):
Outpatient Treatment: Patient recruitment for each outpatient treatment site is shown in Tables 3, 4 and Figure 2. Outpatient treatment was by definition provided by clinicians prior to hospital admission. Outpatient sites included private offices, physician and hospital clinics. Decision to treat (Treatments 1-4; Tables 2A and 2B) was made solely by the physician and patient. All outpatients received a minimum of 200 mg of elemental zinc daily while taking aminoquinolines. Following initial evaluation including PCR testing and initiation of treatment or the decision to provide no treatment, patients returned 3-5 days later for re-evaluation." (Emphasis added)
I'm puzzled how the treating physicians would have known to treat patients with 200 mg of zinc daily? In the protocol document dated April 2020 (still available at clinicaltrials.gov), there is no mention of pre-hospitalization or outpatient treatment. On July 4, 2020, an Appendix G was added that added a prehospitalization step which includes, "Begin immune supportive Rx, including Zn." No quantity of Zn is specified.
Further in a late September YouTube video by the first author he recommended 10 mg of daily zinc for prehospitalized COVID-19 patients.

If the protocol provided to physicians at the outset of the trial did not include prehospitalization instructions, but zinc (of unknown quantity) was included beginning in July, and in late September (after the conclusion of this study) the first author was recommending 10 mg daily Zn... how can the author make the statement that all outpatients received a minimum of 200 mg of elemental zinc daily?
Cheshire pointed out that based on this table in Fleming's preprint, all treatment groups received either 10 mg of zinc orally or 4 mg of zinc intravenously:
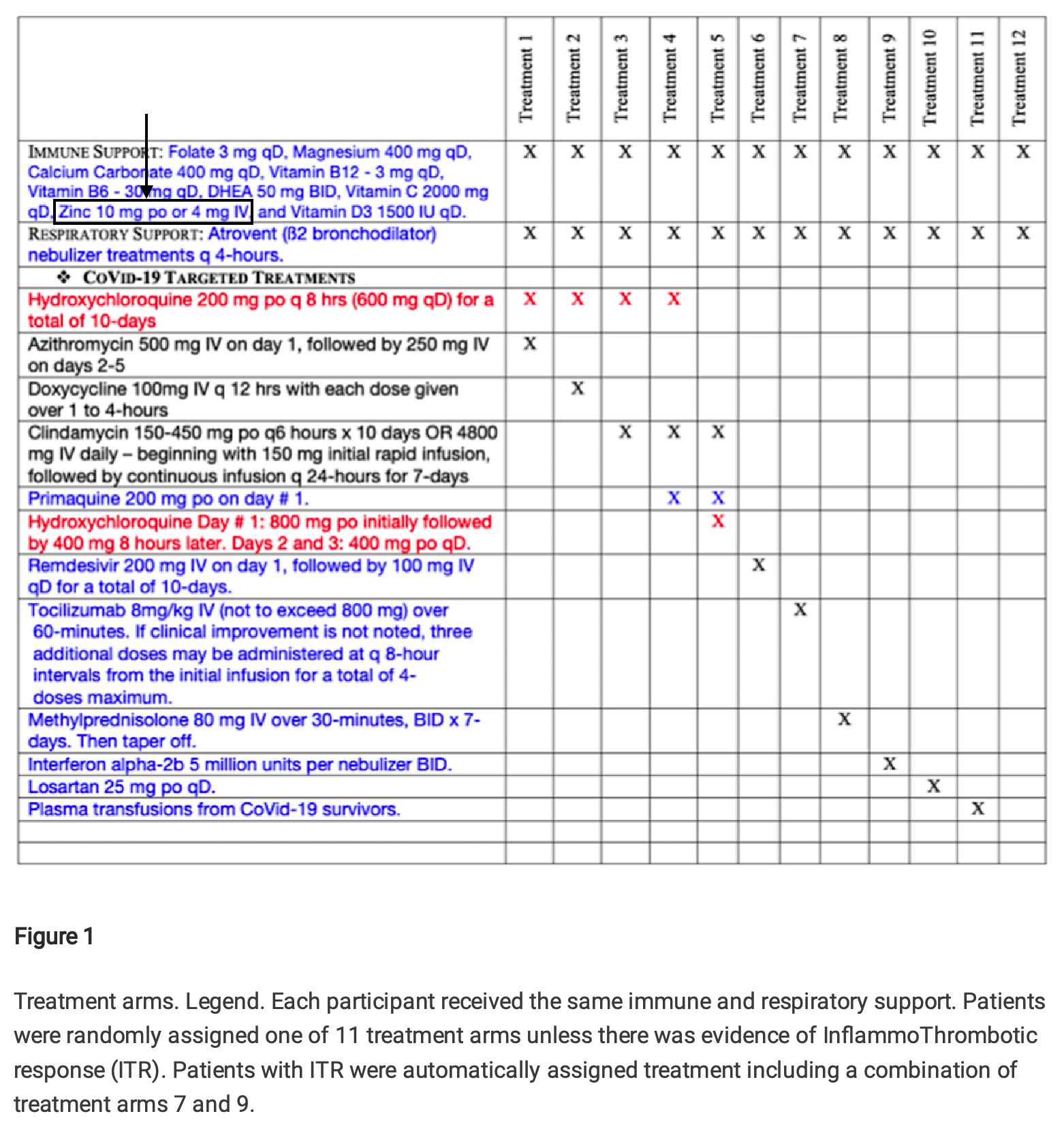
But Fleming responded by saying that his table showed the treatment regime in hospitalized patients and not outpatients:

However in case zinc was suspected to be helpful in treating COVID, why was the dosage of zinc reduced from 200 mg to 10 mg after a patient was hospitalized? And why did Fleming recommend a dosage of only 10 mg of zinc to outpatients in his YouTube video?
The abstract of Fleming's paper said: "The three successful treatment regimens include (1) Tocilizumab & Interferon a-2b, (2) Primaquine, Clindamycin, Tocilizumab & Interferon a-2b, and (3) Methylprednisolone. These three regimens were effective 99.83% of the time and shortened hospital stays from 40 ± 3 days to 1-2 weeks."
It's not clear how he determined the baseline hospital stay of 40 ± 3 days, because his study did not have any control group. I found no other reference to the figure of 40 ± 3 days apart from the abstract. The average duration of 40 days seems far too long, and the standard deviation of 3 days seems too narrow relative to the average duration.
A meta-analysis of COVID hospitalization studies from 2020 said:
"We identified 52 studies, the majority from China
(46/52). Median hospital LoS [length of stay] ranged from 4 to 53 days within
China, and 4 to 21 days outside of China, across 45 studies."
[https://
The conclusion of Fleming's paper said: "Successful treatment interventions focused on (1) avoiding intubation or extubating the patient within a matter of days - less than one week - to minimize the ARDS associated ventilator complications associated with the immunologic ITR to SARS-CoV-2, in addition to (2) using a combination of treatments within the first few days of admission including Interferon a-2b, Tocilizumab, and Methylprednisolone. These combinations were most effective if the patient had already received an aminoquinoline as an outpatient, or Primaquine as an inpatient. When provided the administration of convalescent plasma proved effective; however, given the limited supply of convalescent plasma, the potential consequences of a blood product transfusion including increased potential for thrombosis as a plasma product, and the availability of effective ITR treatments, convalescent plasma should be reserved for cases not responding to Interferon a-2b, Tocilizumab, Methylprednisolone, or the combination of Tocilizumab with Interferon a-2b. These ITR drugs proved most promising when initiated upon admission and when used in combination, reducing hospitalization time from 30-45 days to as little as 18-25 days with 0.17% mortality."
However 18 to 25 days is about 2.6 to 3.6 weeks, and not 1 to 2 weeks like Fleming wrote in the abstract.
The mortality rate of 0.17% seems to match the figure of 99.83% effectiveness that Fleming mentioned in the abstract, so at first I thought that by effectiveness, Fleming simply meant the percentage of subjects who did not die. Hovever then I thought I was wrong, because I noticed that he is supposed to have determined whether a treatment was successful based on three biomarkers: "Successful treatment outcomes were defined using the quantitative measurements of FMTVDM with a reduction of ≥ 25, or a level of < 150, Ferritin levels < 270 ng/ml for men and < 160 ng/ml for women, and an IL-6 level of < 5 pg/ml." But next I noticed that Fleming also wrote: "Three hundred and forty patients entered Phase I and received sequentially added medical Treatment(s) until the patient demonstrated treatment success or expired." So apparently all of his imaginary subjects are supposed to have been treated until they either died or they recovered as measured by one of his three biomarkers, which explains why his efficacy against death is the same as his efficacy determined based on the biomarkers.
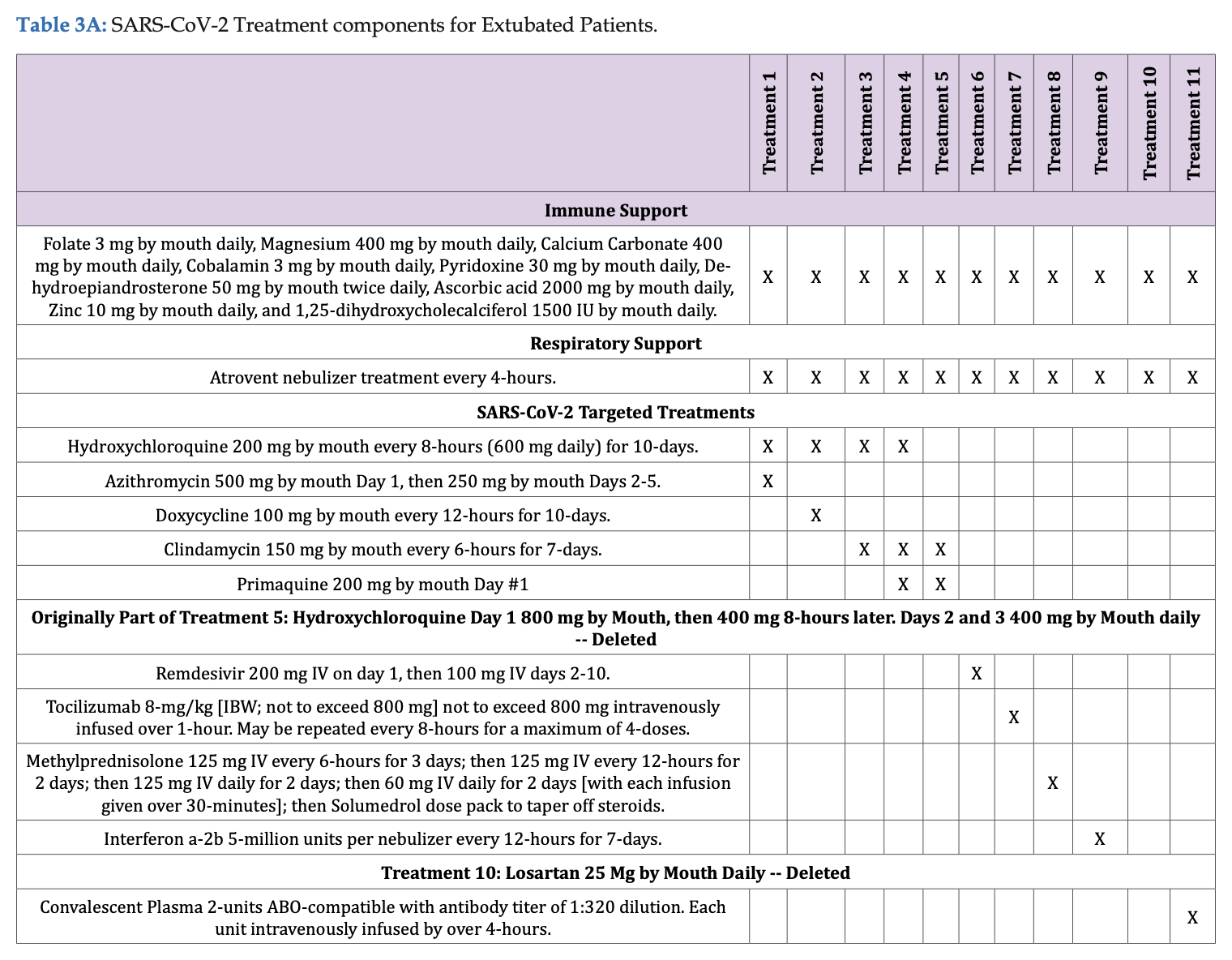
A mortality rate of 0.17% corresponds to one death per a minimum of 572 people (about 0.1748%) and a maximum of 606 people (about 0.1653%). But I think the study didn't even have 572 subjects who received any of Fleming's so-called "three successful treatment regimes". There were only 501 inpatient subjects, and Fleming's paper said: "Outpatient treatment was by definition provided by clinicians prior to hospital admission. Outpatient sites included private offices, physician and hospital clinics. Decision to treat (Treatments 1-4; Tables 3A & 3B) was made solely by the physician and patient." And Table 3A shows that treatments 1-4 didn't include any of Fleming's "three succesful treatment regimes", even though clindamycin alone was given to some outpatients (in the imaginary realm where Fleming conducted his trial):
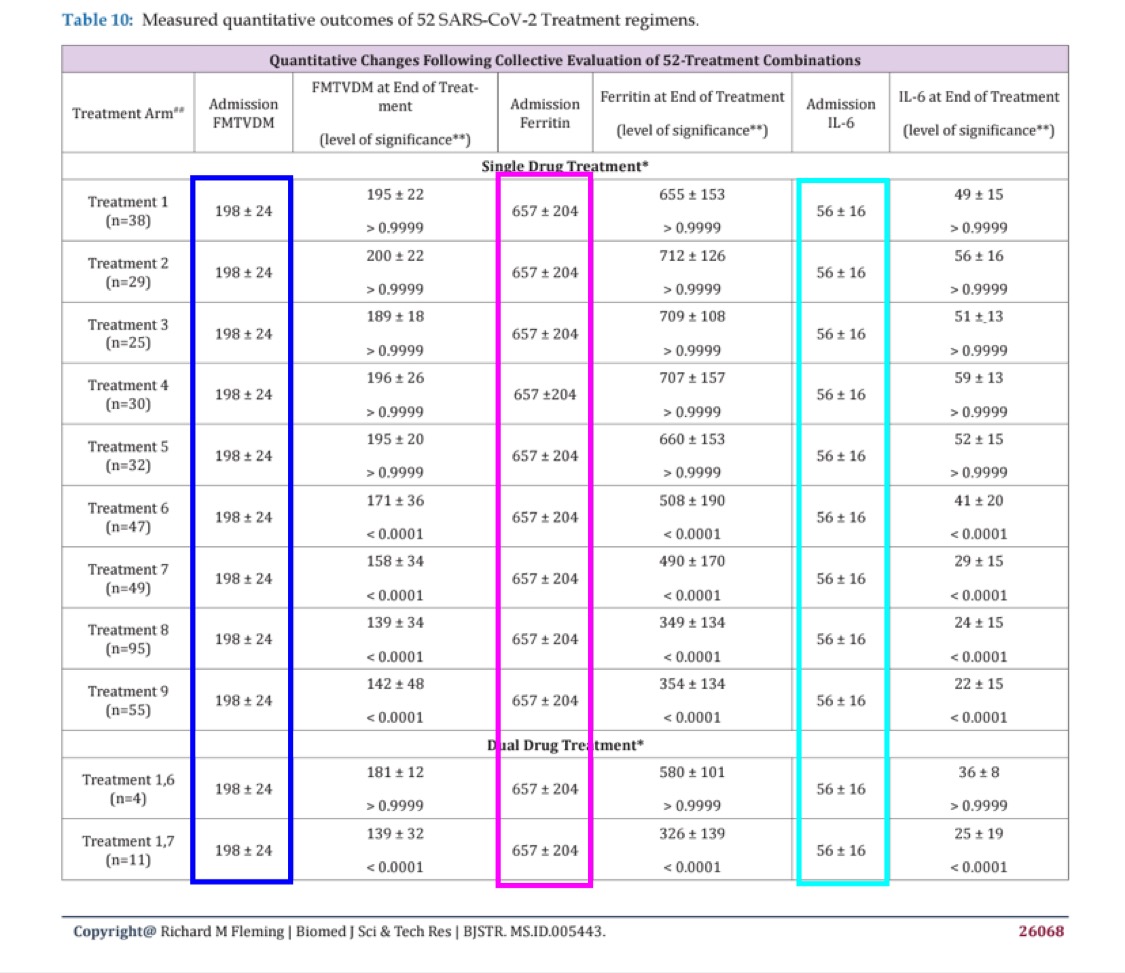
Cheshire pointed out that people in all 11 treatment arms had the same baseline levels of ferritin and IL-6 at admission:
When I asked Fleming to explain it, he replied: "As explained in the paper, although clearly not read or
comprehended by others, all participants began at baseline. This was
baseline. They were then randomly assigned to treatments and then based
upon measured FMTVDM outcomes, additional treatments were added or not.
Statistical analysis was by multiple ANOVA." [https://
But I still don't understand how it's possible for each arm to have the same levels of ferritin and IL-6 at admission.
Someone at Retraction Watch posted this comment: [https://
According to the trial registration, "study chair" Fleming is conducting a clinical trial testing FDA regulated drugs for diagnosis and treatment in patients with Covid-19. Since there are no approved treatments for Covid-19, and IND application to FDA is required.
Per the FDA debarment notice, Fleming is prohibited "for 10 years from providing services in any capacity to a person that has an approved or pending drug product application."
https://
www. govinfo. gov/ app/ details/ FR- 2018- 09- 28/ 2018- 21210 Perhaps Retraction Watch could clarify with FDA that the debarment includes provision of services in FDA regulated research.
Cheshire posted this reply:
Source: https://
www. healthgrades. com/ media/ english/ pdf/ sanctions/ H G P Y A 5 9 E 0 F 8 B 7 C 7 4 4 E C B B 0 5 2 1 2 0 1 0 . According to this State of Nebraska "Petition for Disciplinary Action," Fleming is "permanently excluded from Medicare, Medicaid, Tricare, and all other federal healthcare programs."
I verified that he is on the exclusion list accessible here: https://
oig. and that site says:hhs. gov/ exclusions/ index. asp OIG has the authority to exclude individuals and entities from Federally funded health care programs for a variety of reasons, including a conviction for Medicare or Medicaid fraud. Those that are excluded can receive no payment from Federal healthcare programs for any items or services they furnish, order, or prescribe. This includes those that provide health benefits funded directly or indirectly by the United States (other than the Federal Employees Health Benefits Plan).
OIG maintains a list of all currently excluded individuals and entities called the List of Excluded Individuals/Entities (LEIE). Anyone who hires an individual or entity on the LEIE may be subject to civil monetary penalties (CMP). To avoid CMP liability, health care entities should routinely check the list to ensure that new hires and current employees are not on it.
Based on this, it sounds unlikely that Fleming/Dr. who? would be in a position to legally run a clinical trial.
However I don't know to what extent US regulations would've applied to Fleming's trial since none of his imaginary trial sites were located in the United States. There were 5 sites in India, 4 in Germany, 4 in Brazil, 3 in Philippines, 3 in Cuba, 3 in Belgium, and 1 in South Africa.
A blog post about the journal where Fleming's paper was published
said: [http://
Although the ISSN entry for the Biomedical Journal of Scientific & Technical Research identifies it as a US publication, its mangled management of the English language suggests otherwise. The website describes the goal of this journal's "publishers" as follows:
The only motto of Biomedical Journal of Scientific & Technical Research (BJSTR) Publishers is accelerating the scientific and technical research papers, considering the importance of technology and the human health in the advanced levels and several emergency medical and clinical issues associated with it, the key attention is given towards biomedical research. Thus, asserting the requirement of a common evoked and enriched information sharing platform for the craving readers.
BJSTR is such a unique platform to accumulate and publicize scientific knowledge on science and related discipline. This multidisciplinary open access publisher is rendering a global podium for the professors, academicians, researchers and students of the relevant disciplines to share their scientific excellence in the form of an original research article, review article, case reports, short communication, e-books, video articles, etc.
BJSTR Publishers are self supporting, with no dependency on any other external sources (like universities, centers) for funds and strives for the best and enhanced quality publications competes the world wide open access publishing market.
We always rely on the support from the members of our BJSTR family that is relevantly our Authors, Editorial Committee members, advisory board, Reviewers Board and all the technical support teams all over the globe. We trust in the reciprocated coordination and cooperation in terms of sharing the scientific knowledge of individuals and Groups of Research centers/areas will in turn educates and provokes in advanced researches. In this case we would like to act as a media that anchors in the transformation of information in the form of global online publication.
With writing like this, the absence of "dependency on any ... external sources (like universities, centers) for fund" is understandable. So is the fact that the journal is not in the National Library of Medicine collection and not indexed for Medline.
When one researcher "submitted a string of machine-generated nonsense" entitled "The Expression of the Proper-Name Effect Reinforces the Disarticulation ofCommunicative [sic] Rationality," the article was "quickly accepted after scrupulous peer review." Evidently, the editors did not notice that the author was Harold E. Potter of the Institute of Improbabilistics, University of Bogus, UK. An author posting on Researchgate reports that when he declined to pay the $600 fee, the journal dropped the price to $99.
Wisely, the publishers do not reveal their identities. The domain name biomed-res.org in the email is registered to a privacy service. The contact information on the website is Biomedical Research Network+, LLC, 1 Westbrook Corporate Center, Suite 300, one Westchester, IL 60154, USA +1 (720) 414-3554 Fax - (720) 367-5187 info@biomedres.us angelaroy@biomedres.us. The registrant of biomedres.us provides the same physical address along with the name Biomedical Research Network + LLC and email URL of openaccessnetworks@gmail.com. The Westbrook Corporate Center is a property with "virtual office" space on sale for as little as $2 a day.
Someone posted this comment to the blog post: "ALL these BJSTR, IRIS, CRIMSON, LUPINE are interconnected
to SRAWIK GROUP a proprietor company in Hyderabad India". However
I'm not sure if it's correct or not, because there were no other hits on
Google when I searched for
srawik "biomedical journal of scientific".
The associate editor of the BJSTR journal is suposed to be someone
called "Angela Roy", whose location at
Twitter is listed as "600 Third Avenue, 2nd floor,
New York - 10016, USA". [https://
This section addresses the PDF linked at the top of this page:
https://
Fleming wrote: "When the EUA documents were used
for the statistical analysis of the Pfizer, Moderna, and Janssen Drug
Vaccine Biologics, and the Chi-Square analysis of the results published
in those EUA documents was analyzed for the Pfizer vaccine as shown in
the following graphic, there was no statistical difference between
vaccinated and un-vaccinated people diagnosed with having COVID-19. To
be statistically different (a benefit for people
being vaccinated) the 'p (probability)- value' must be less than or
equal to less than 5 times per hundred people. This is the scientific
definition of statistical benefit and is written as 'p<0.05'. In the graphic the p-value was
0.224418 [sic; actually 0.244218] and is NOT
statistically significant; i.e. there is no statistical difference in
the number of people diagnosed with COVID who were vaccinated when
compared with the non-vaccinated group of people." And he showed
the image below, where the table on the left is from a review memorandum
for the emergency use authorization of the Pfizer vaccine. [https://

There were 8 cases out of 17,411 people in the vaccine group and 162 cases out of 17,511 people in the placebo group, so common sense should've told Fleming that the difference between the groups was statistically significant, and he should've noticed that there was something wrong with his calculation because he only got a p-value of about 0.24.
In Fleming's contingency matrix, the "observed" column shows the number of people with
no case, or for example 17411- for Pfizer. But I
don't understand how he calculated the "expected" column. The numbers in parentheses show
the result of rowSums( (for example 34652* for
the top left square).
However the proper way to make the contingency matrix would've been to have one column for the number of people with a COVID case and another column for the number of people with no COVID case. It gave me a p-value of about 9e-32 here:
>cases=c( 8, 162); people=c( 17411, 17511) > m=cbind( case=cases, no_ case=people- cases) > rownames( m) =c( " Pfizer", " placebo") > m case no_ case Pfizer 8 17403 placebo 162 17349 > chisq. test( m) # p- Pearson'value shown as `< 2. 2e- 16` due to limit of floating point precision s Chi- squared test with Yates' continuity correction data: m X- squared = 137. 5, df = 1, p- value < 2. 2e- 16 > .Machine$ double. eps # smallest positive float `x` [where `1+ x! =1` 1] 2. 220446e- 16 > chisq. test( m)$ p. value # approximation of actual p- [value 1] 9. 395097e- 32 > expected=rowSums( m)%*% t( colSums( m))/ sum( m) > expected case no_ case [ 1,] 84. 7566 17326. 24 [ 2,] 85. 2434 17425. 76 > sum(( m- expected)^ 2/ expected) # manual calculation without correction [1] 139. 3046 > chisq. test( m, correct=F)$ stat # matches manual calculation X-squared 139. 3046
Kevin McCairn also got a similar p-value when he used MATLAB to do a
chi-squared test (but the reason why McCairn said he
got a lower p-value than me was because my p-value was shown as
< by R even though it was actually about 9e-32
which was similar to McCairn's result). McCairn wrote: [https://
Richard I'm not following the logic, not that I trust Pfizer data, but the appropriate use of the Chi-squared in the tables above would be calculated as follows (MATLAB code below), I get a smaller P Value than Henjin, but I did no post-hoc corrections as it's only a 2x2 contingency table, and the result is highly significant.
%Data observed = [8, 162; 17403, 17349]; % Observed frequencies total = sum( observed, ' all'); row_ totals = sum( observed, 2); col_ totals = sum( observed, 1); % Expected frequencies expected = (row_ totals * col_ totals) / total; % Chi- squared calculation chi_ squared = sum(( observed - expected).^ 2 ./ expected, ' all'); % Degrees of freedom df = (size( observed, 1) - 1) * (size( observed, 2) - 1); % P- value calculation p_ value = 1 - chi2cdf( chi_ squared, df); % Critical value for p = 0. 05 critical_ value = chi2inv( 0. 95, df); % Display results fprintf( ' Chi- squared statistic: %. 2f\ n', chi_ squared); fprintf( ' Degrees of freedom: %d\ n', df); fprintf( ' Critical value (p=0. 05): %. 2f\ n', critical_ value); fprintf( ' P- value: %g\ n', p_ value); % Use %g for full precision scientific notation % Conclusion if chi_ squared > critical_ value disp( ' The result is significant at p = 0. 05. '); else disp( ' The result is not significant at p = 0. 05. '); end The result is significant at p = 0. 05 χ2=139. 3046 P=7. 89×10−32
Fleming wrote "No other Coronavirus contains a Furin Cleavage site as shown in the following graphic":

But his graphic only contained sarbecoviruses, and not other types of coronaviruses.
Many merbecoviruses like MERS and HKU5 have a furin cleavage site at the S1/S2 junction. The closest relative of sarbecoviruses are hibecoviruses, which are a small subgenus of betacoronaviruses that I believe currently includes only 4 published virus sequences, which are Hp-betacoronavirus/Zhejiang2013, Zaria bat coronavirus ZBCoV, CD35, and CD36. But all of them except ZBCoV have an FCS at the S1/S2 junction:

Feline infectious peritonitis virus is also a coronavirus, but there
are sequences of FIPV that have an FCS at the S1/S2 junction with the
residues PRRAR:

Fleming wrote: "The odds of such a cleavage site occurring spontaneously (naturally) is 3.21 x 10-11. [...] Simply put, the Furin cleavage site 'critical' for the SARS-CoV-2 viruses' entry into human cells resulting in disease and death, is astronomical. It has it not found in any other coronavirus at the critical S1/S2 location, and the US Government which has funded the Gain-of-Function research, owns the patent for this Furin Protease Cleavage Site, also associated with the HIV glycoprotein (HIV gp120) 120 (sialic acid raft receptor) and cancer progression."
The figure of 3.21e-11 comes from a paper by Ambati et al. titled
"MSH3 Homology and Potential Recombination Link to
SARS-CoV-2 Furin Cleavage Site". [https://
The paper by Ambati et al. included the image below with this caption: "Calculations of the probability of natural occurrence of the 19nt sequence under study. The SARS-CoV-2 genome is ~30,000 nucleotides long (P1). The patented sequence is ~3,300 nucleotides long (P2). The patented library encompasses 24'712 sequences of varying lengths with median length being in the range of 3,300 nucleotides. Conventional probability calculations are given of the probability of the presence of a 19-nucleotide sequence in the human genome and in one of the patented library sequences."

Ambati calculated the likelihood that a given 19-base segment would
be included in SARS-CoV-2, and he multiplied it with the likelihood that
the same segment would be included in the single Moderna patent that
happened to match the 19-base segment. But he should've instead just
calculated the likelihood that a given 19-base segment would match the
Moderna patent by chance, like what he did in his P2c
probability, or the likelihood that a given 19-base segment would match
any Moderna patent published before 2020.
In order to search the patent BLAST database for sequences that were
published before 2020 and that matched the keyword "Moderna", I used the Entrez query
0:. It returned a total of 669,810 sequences
with a total length of about 1,011,731,339 bases. So if all sequences
are assumed to be random, then the likelihood that any of them would
match a given 19-base segment would be about 0.007:
1-(( (where the
likelihood of one or more matches is calculated by subtracting the
likelihood of zero matches from 1, and 1-. is the
likelihood that a single 19-base segment doesn't match the target
sequence, and 1e9* is the approximate number of 19-base
segments on both strands in all Moderna patent sequences). But
since there's 8 different ways to choose a 19-base segment of SARS-CoV-2
that contains the FCS insert and a total of 7 surrounding bases from
either side combined, you can further multiply the likelihood by 8.
The journal that published Ambati's paper later published a response
to his paper, which pointed out these problems with his probability
calculation: [https://
The 19-base subsequences of a longer sequence are not fully independent from each other because of overlap, which can be taken into account by using a Markov chain model, even though it doesn't make much difference in the results of the probability calculation.
Ambati's P2c probability indicated the likelihood
that a given 19-base segment would occur exactly once among the patent
sequences. He should've instead calculated the likelihood that the
19-base segment would occur one or more times among the patent
sequences, which is the same as one minus the likelihood of zero
occurrences: 1-(( (where 1-. is the likelihood that a
segment at a given position of a given patent sequence does not match
the 19-base target sequence, and ( is the
number of 19-base segments on a single strand of all patent
sequences). The resulting probability is about 0.000295014, which
is only about 0.01% higher than Ambati's P2c probability,
so it doesn't make much difference though.
Ambati forgot to take into account that the match can occur on
either strand, so he should've multiplied the P2c
probability by 2.
Ambati's probability should've been multiplied 8, because there's 8 different ways to select a 19-base segment that contains the 12-base FCS insert and 7 surrounding bases.
Ambati et al. said that the sequence listing included in the 5 Moderna patents contained a total of 24,712 sequences, but I don't know where they got the figure, because actually the sequence listing contains 33,915 sequences, out of which 4,573 are amino acid sequences and 406 are RNA sequences that contain U bases. The remaining number of DNA nucleotide sequences is 28,936, which doesn't match Ambati's number of 24,712 sequences. Ambati guessed that the median length of the sequences was about 3,300 bases, but it doesn't match my calculation, where the median length was 1,250.5 bases when I excluded the amino acid and RNA sequences.
But anyway, Fleming was wrong to say that "the odds of such a cleavage site occurring spontaneously (naturally) is 3.21 x 10-11". The likelihood that the FCS would occur spontaneously is not the same as the likelihood that a specific Moderna patent would happen to match the specific insert that happened to add the FCS along with 7 surrounding bases. There are also many other possible inserts that could've added an FCS to SARS-CoV-2, and even many combinations of point mutations that could've added an FCS. The FCS sequence only needs to have two arginine residues with two other residues in between, and the sequence can be located at multiple different spots near the S1/S2 junction.
There are 6 different codons for arginine, so there are many
different ways to introduce mutations that produce an arginine residue.
For example in RaTG13, the codons after the spot where Wuhan-Hu-1 has
RRAR are S:, but it would
change to RVAR if the 3rd nucleotide was changed to
A or G and the 12th nucleotide was changed to
A or G.
The codons around the S1/S2 junction in RaTG13 are
T:, which would match
RXXR if either the T at the start or the
A at the end changed to R. But there's 3
combinations of 2 nucleotide changes and 3 combinations of 3 nucleotide
changes which would change the T at the start to
R, and there's 1 combination of 2 nucleotide changes and 5
combinations of 3 nucleotide changes which would change the
A at the end to R.
The likelihood that the 12-base FCS insert would occur by chance is not even the same as the likelihood that the 12-base FCS insert and the 7 surrounding bases would occur by chance. And the likelihood that the specific FCS motif in Wuhan-Hu-1 would occur by chance is not the same as the likelihood that any FCS motif would occur by chance. And the likelihood that the FCS insert in Wuhan-Hu-1 would occur naturally is not the same as the likelihood that it would occur by chance, because certain mutations might be favored by natural selection. And the likelihood that the 19-base segment happened to match one specific Moderna patent is not the same as the likelihood that the 19-base segment would have occurred naturally.
In order to estimate the likelihood that the 12-base insert and a
total of 7 surrounding bases from either side would have an exact match
in the patent database, you can go here:
https://A, which
consists of the 12-base FCS insert and 7 surrounding bases from either
side. Then switch the database to patent sequences, insert "SARS-CoV-2 (taxid:2697049)" as the organism and click
the exclude checkbox, and under algorithm parameters set maximum results
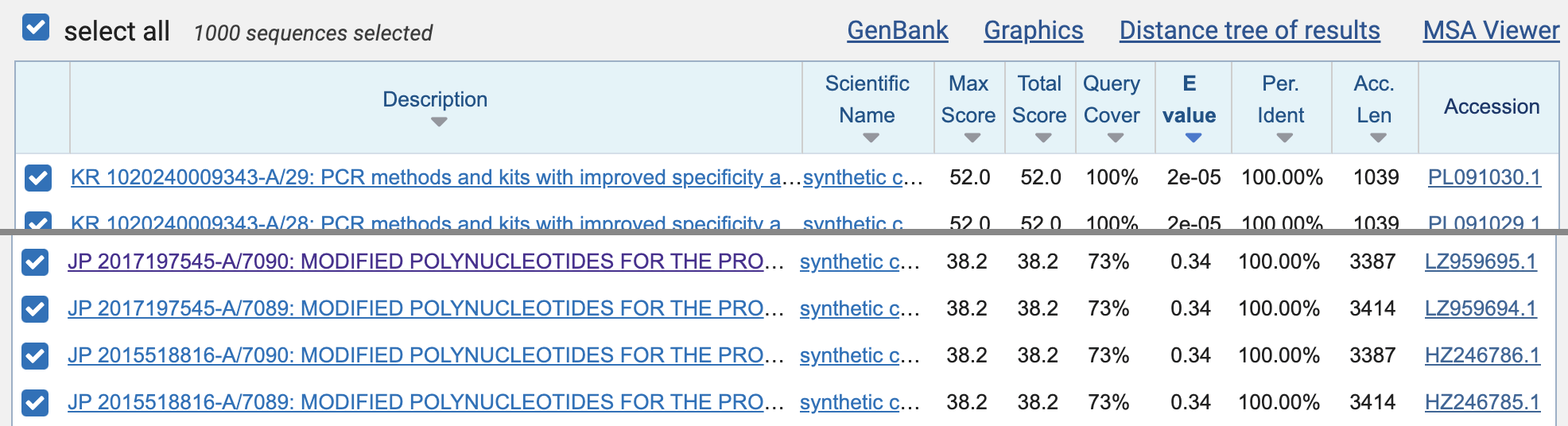
to 1000, and then click "BLAST". When I ran
the query, the first 392 matches were mostly matches to SARS-CoV-2 that
were not removed by the species filter, but the matches to the Moderna
patent sequences were listed on rows 393 to 396 (JP
2017197545-A/7090, JP 2017197545-A/7089, JP 2015518816-A/7090, and JP
2015518816-A/7089). However the E-value of all matches was about
0.34, which means that about 0.34 similarly close or closer matches were
expected to occur by chance in the patent database:
The E-value depends on the size of the database, and the patent database is fairly small compared to other BLAST databases like the core nucleotide database. So when I repeated the previous search but I switched to the core nucleotide database, there were a bunch of bacterial sequences that got a perfect match to some 19-base subsegment of the 33-base query, but the E-values of the matches were about 8.9, which means that about 8.9 similarly close or closer matches were expected to occur by chance.
As an indication of how likely 19-base matches are to occur by chance, there are 29,885 different 19-base segments of Wuhan-Hu-1, but 1,670 or about 6% of them had an exact match to the GRCh38 human reference genome:
$brew install bowtie2 seqkit $ wget ftp:// ftp. ccb. jhu. edu/ pub/ data/ bowtie_ indexes/ GRCh38_ no_ alt. zip; unzip GRCh38_ no_ alt. zip $ curl -s ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=fasta& id=MN908947' > sars2. fa $ seqkit seq -s sars2. fa| awk -F ' ' '{ for( i=1; i< length- 18; i++) print " > " i"\ n" substr($ 0, i, 19)} ' > frag. fa $ bowtie2 -p4 -- no- unal -x GRCh38_ noalt_ as/ GRCh38_ noalt_ as -- score- min C, 0,- 1 -fU frag. fa> temp 29884 reads; of these: 29884 (100. 00%) were unpaired; of these: 28214 (94. 41%) aligned 0 times 1427 (4. 78%) aligned exactly 1 time 243 (0. 81%) aligned > 1 times 5. 59% overall alignment rate
Ambati's 19-base segment matches bases 23601 to 23619 of Wuhan-Hu-1,
which consists of the codons for PRRAR and 2 surrounding
bases on from both sides. The 19-base segment matches the 12-base
insertion in Wuhan-Hu-1 and the next 7 bases. Jikkyleaks has made a huge
deal about Ambati's BLAST match, which he has dubbed "Modernagate". His theory is that SARS-CoV-2
acquired the 12-base insert in a cell culture from the human MSH3 gene
through an accidental recombination event, so that the 7 flanking bases
after the insert somehow helped facilitate the accidental recombination.
His theory doesn't make any sense, but even if it did, it's not clear if
the 7-base flanking region after the insert would've helped to
facilitate accidental recombination much better than a 6-base or 5-base
flanking region. And if the last two bases from Jikky's segment are left
out so the flanking region is only 5 bases long, then the 17-base
segment has a perfect match to all of these patent sequences published
before 2020:
$curl ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=xml& id=HC903376, HC915932, HC908933, HC905783, HB443296, GN095906, BD179519' > temp. xml $ xmlstarlet fo -D temp. xml| xml sel -t -m // GBSeq -v GBSeq_ accession- version -o \| -v GBSeq_ definition -o \| -v GBSeq_ create- date -o \| -v .// GBReference/ GBReference_ title -o \| -v .// GBReference/ GBReference_ journal -o \| -n| awk -F\| '$ 3!~/ 202/ ' HC903376. 1| Sequence 13528 from Patent EP2194140| 18- JUN- 2010| Process for the production of fine chemicals| EP2194140- A2 13528 09- JUN- 2010 Metanomics GmbH (DE)| HC915932. 1| Sequence 26085 from Patent EP2194140| 18- JUN- 2010| Process for the production of fine chemicals| EP2194140- A2 26085 09- JUN- 2010 Metanomics GmbH (DE)| HC908933. 1| Sequence 19086 from Patent EP2194140| 18- JUN- 2010| Process for the production of fine chemicals| EP2194140- A2 19086 09- JUN- 2010 Metanomics GmbH (DE)| HC905783. 1| Sequence 15936 from Patent EP2194140| 18- JUN- 2010| Process for the production of fine chemicals| EP2194140- A2 15936 09- JUN- 2010 Metanomics GmbH (DE)| HB443296. 1| Sequence 19 from Patent WO2009077406| 14- JUL- 2009| Lipid metabolism proteins, combinations of lipid metabolism proteins and uses thereof| WO2009077406- A1 19 25- JUN- 2009 BASF Plant Science GmbH (DE)| GN095906. 1| Sequence 687 from Patent WO2009037279| 16- APR- 2009| Plants with increased yield| WO2009037279- A1 687 26- MAR- 2009 BASF Plant Science GmbH (DE)| BD179519. 1| Highly thermophilic bacterium- derived protein and gene encoding it| 15- MAY- 2003| Highly thermophilic bacterium- derived protein and gene encoding it| JP2002325574- A 10 12- NOV- 2002 THE INSTITUTE OF PHYSICAL AND CHEMICAL RESEARCH|
To reproduce the output of code above, go here:
https://CTCCTCGGCGGGCACGT to the field at the top, switch the
database to "Patent sequences(pat)", and click "BLAST". Then copy the accession numbers up to the
last result where query coverage and identity are both 100%, and run the
code above with the accession IDs joined by commas inserted as the id
parameter.
All 5 Moderna patents have the same sequence listing that contains a
total of 33,915 sequences. It's unusual for a single patent to include
so many sequences, so McKernan said the patents were "kitchen sink patents". You can download all
sequences from here:
https://
$unzip US09587003- 20170307- SUPP. zip $ sed ' s/.* SEQUENCE: />/;/^[ < ]/ d; s/ *[ 0- 9]*\ r$//; s/ // g' Psips/ Data/ 03/ US/ 2017/ 870/ 095/ B2/ sequence/ US09587003- 20170307- S00001. TXT| seqkit stat file format type num_ seqs sum_ len min_ len avg_ len max_ len moderna. fa FASTA DNA 33, 915 57, 673, 267 7 1, 700. 5 18, 787
The sequence listing contains 4,573 amino acid sequences and 406 RNA sequences. And out of the remaining 28,936 sequences, 24,363 sequences don't have any stop codons in the middle if they are translated:
$tr -d \\ r< Psips/ Data/ 03/ US/ 2017/ 870/ 095/ B2/ sequence/ US09587003- 20170307- S00001. TXT| awk '/ TYPE: DNA/ ' RS= ' ORS=\ n\ n'| sed ' s/ < 400> SEQUENCE: />/;/^[ < ]/ d; s/ *[ 0- 9]*$//; s/ // g'| seqkit grep -svp u> moderna. fa $ seqkit translate moderna. fa| seqkit grep -svrp'\*. '| seqkit stat file format type num_ seqs sum_ len min_ len avg_ len max_ len - FASTA Protein 24, 363 12, 482, 339 3 512. 3 5, 891
When I generated all 19-base segments of Wuhan-Hu-1 with the poly(A) tail removed, and I searched for the segments within the sequence listing in the Moderna patents, there were 3 other 19-base segments besides Jikky's segment that had a perfect match to the patent sequences:
$bowtie2- build -- thread 4 moderna. fa{,} $ seqkit replace -sip' a*$ ' sars2. fa| seqkit sliding -W19 -s1| seqkit replace -p'.*: ' > frag. fa $ bowtie2 -p4 -- no- unal -x moderna. fa -- score- min C, 0,- 1 -fU frag. fa> moderna. sam $ (echo sars2_ pos patent_ sequence_ number match_ start_ in_ patent_ sequence sequence; grep -v ^@ moderna. sam| cut -f1, 3, 4, 10)| column -t sars2_ pos patent_ sequence_ number match_ start_ in_ patent_ sequence sequence 3180- 3198 18510 967 AAGAAGAGCAAGAAGAAGA 4459- 4477 26445 1172 AGTTTCAACTATACAGCGT 8293- 8311 30068 1160 CTATAACAAAGTTGAAAAC 23601- 23619 11651 2760 CTACGTGCCCGCCGAGGAG
If the amino acid and RNA sequences are excluded, the Moderna patents
have a total of about 49 million 19-base segments. And there's 29,885
19-base segments in Wuhan-Hu-1. So if both collections of segments would
be random, the expected number of exact matches between the collections
would be ., which is about 11.
Jikky wrote about his 19-base segment that "In
order for that sequence to have arisen in that virus, the virus which
was manufactured with its HIV inserts, had to have had been infected
into patented cell lines supplied by Moderna that had that unique
sequence not seen in any other virus." [https://
Jikky stated it as a fact that SARS-CoV-2 was made using an MSH3 cell
line patented by Moderna, because he wrote: "Irrespective of whether it was released there it
distracts from the fact that it was made in a lab using a Moderna
patented MSH3_mut cell line." [https://
Kevin McKernan was blocked by Jikky in 2022 after McKernan debunked
Modernagate in this thread:
https://
Added later: Modernagate was now also debunked here by the Japanese
patent expert Patent_SUN:
https://
The following five patent publications ① to ⑤ each includes the above reverse complementary sequence in its Sequence Listing, but no patent rights have been granted to the reverse complementary sequence itself.
①US9149506B2
②US9216205B2
③US9255129B2
④US9301993B2
⑤US9587003B2Therefore, we must carefully explain the reverse complementary sequence so as not to mislead the public. Regarding those patent publications ① to ⑤, by referring to the "We claim" section (scope of Claims) attached to the end of each specification, we can understand where the patent rights exist.
As we can see in each "We claim" section of the patent publications ① to ⑤, there is no description of "CTACGTGCCCGCCGAGGAG."
From this fact, we can understand that the patent rights have NOT been granted to it.
[...]
The topic has strayed off course, but realistically, it is not possible for Moderna to obtain a patent for this specific sequence, "CTACGTGCCCGCCGAGGAG," or just a part of it (i.e., the furin cleavage site). This is because Moderna disclosed "SEQ ID 11652" as a complete sequence, which includes that specific sequence, but did not disclose the segment as an independent invention. This fails to meet the support requirement for patentability, and it is unlikely that a patent will be granted for this specific sequence in the future.
Furthermore, even if Moderna were to attempt to obtain a patent right for "SEQ ID 11652," I believe it would be a difficult process. Realistically, if Moderna were to pursue a patent right for "SEQ ID 11652," the specification would need to explicitly describe the usage, function, and effects of "SEQ ID 11652" in order to meet the aforementioned support requirement. However, none of the specifications in the Patents 1 to 5 mention "SEQ ID 11652," nor is there any disclosure suggesting it.
Additionally, Moderna does not address the complementary sequence of "CTACGTGCCCGCCGAGGAG," which is "CTCCTCGGCGGGCACGTAG," anywhere in the specification. Therefore, the patent rights of the Patents 1 to 5 never extend to the 'CTCCTCGGCGGGCACGTAG' sequence of the COVID-19 virus.
Another addendum: I went to nucleotide BLAST:
https://0:. Then under algorithm
parameters, I reduced the word size to 16, increased the expect
threshold to 1000, and increased maximum target sequences to 1000.
There were a total of 495 results. The best matches with 100% identity were 23 bases long, but even they had an expect value of about 2.3, which means that about 2.3 similarly close or closer matches were expected to occur by chance:
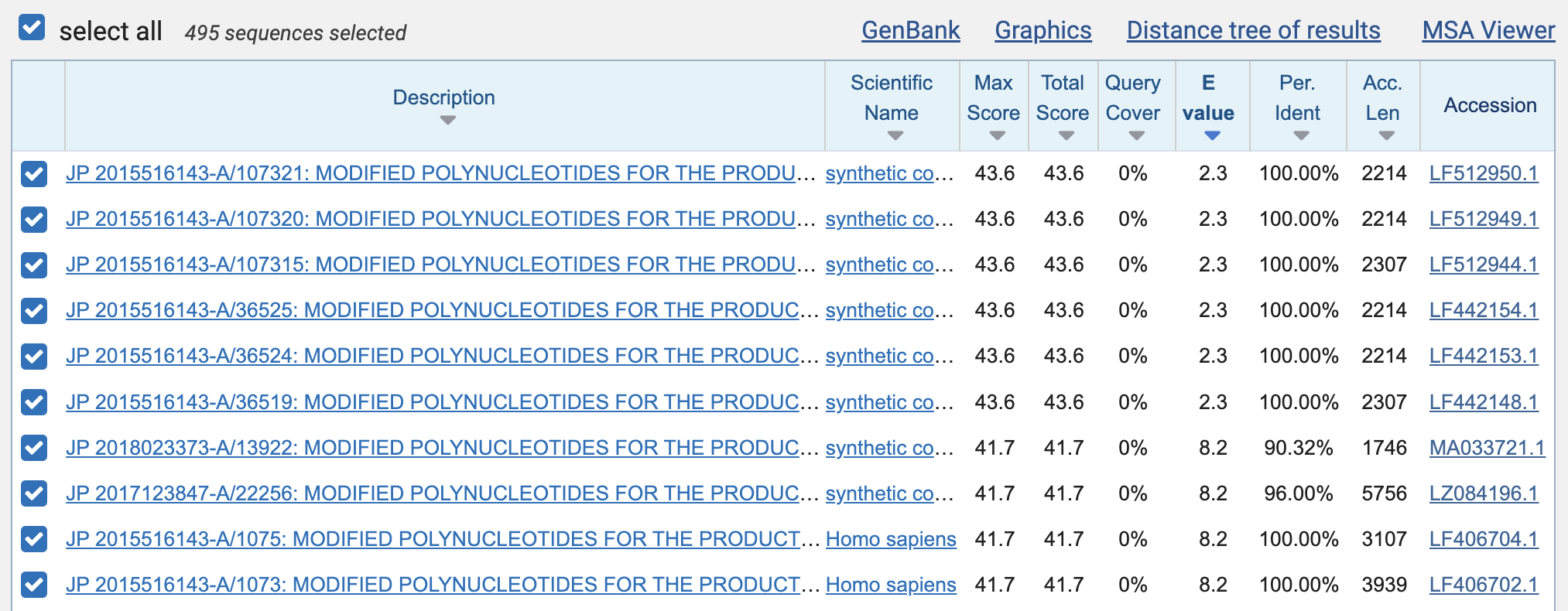
The expect values of the perfect 19-base matches were about 381, which means that about 381 similarly close or closer matches were expected to occur by chance. (And that wasn't even in the whole patent database, but only within the set of patent sequences that were published before 2020 and that matched the word "Moderna".)
Then in order to download a TSV file of the results, I clicked "Download" and selected "Hit Table (text)". The TSV file shows that there were 176 perfect matches with a length of 19 bases, 55 with length 20, 6 with length 21, 13 with length 22, and 6 with length 23:
$awk '$ 3==100& &$ 4> =19{ a[$ 4]++} END{ for( i in a) print i, a[ i]} ' Downloads/ XRM91BX9013- Alignment. txt 19 176 20 55 21 6 22 13 23 6
The perfect matches that were at least 19 bases long came from 256 unique patent sequences:
$awk '$ 3==100& &$ 4> =19' Downloads/ XRM91BX9013- Alignment. txt| cut -f2| sort -u| wc -l 256
Among the perfect matches that were at least 19 bases long, there weren't even too many duplicate matches, because the matches started at 92 unique positions of the genome of SARS-CoV-2, and they covered 1,532 bases of the genome of SARS-CoV-2 which is about 5% of the total length of the genome:
$awk '$ 3==100& &$ 4> =19' Downloads/ XRM91BX9013- Alignment. txt| cut -f7| sort -u| wc -l 92 $ awk '$ 3==100& &$ 4> =19{ for( i=$ 7; i< =$ 8; i++) a[ i]} END{ print length( a)} ' Downloads/ XRN2HBFD016- Alignment. txt 1532
When I clicked "Search Summary", it showed
that there were a total of about 700,000 sequences in the patent
database that matched the Entrez query 0:,
and their total length was about a billion bases:
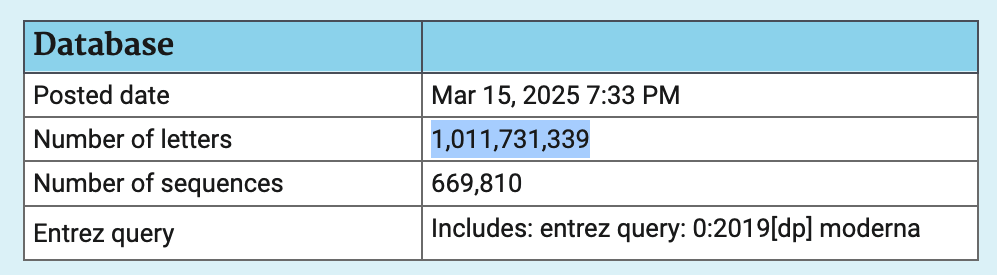
The total length of all sequences in the patent database in March 2025 was about 28.3 billion bases. So about 4% of the total length was made up by sequences that were published before 2020 and that matched the keyword "Moderna".
The next plot shows all matches with the same or lower expect value as the match to Jikky's 19-base segment, where the matches on the top row are similarly close as Jikky's match, and the matches on the 4 rows below it are closer than Jikky's match:
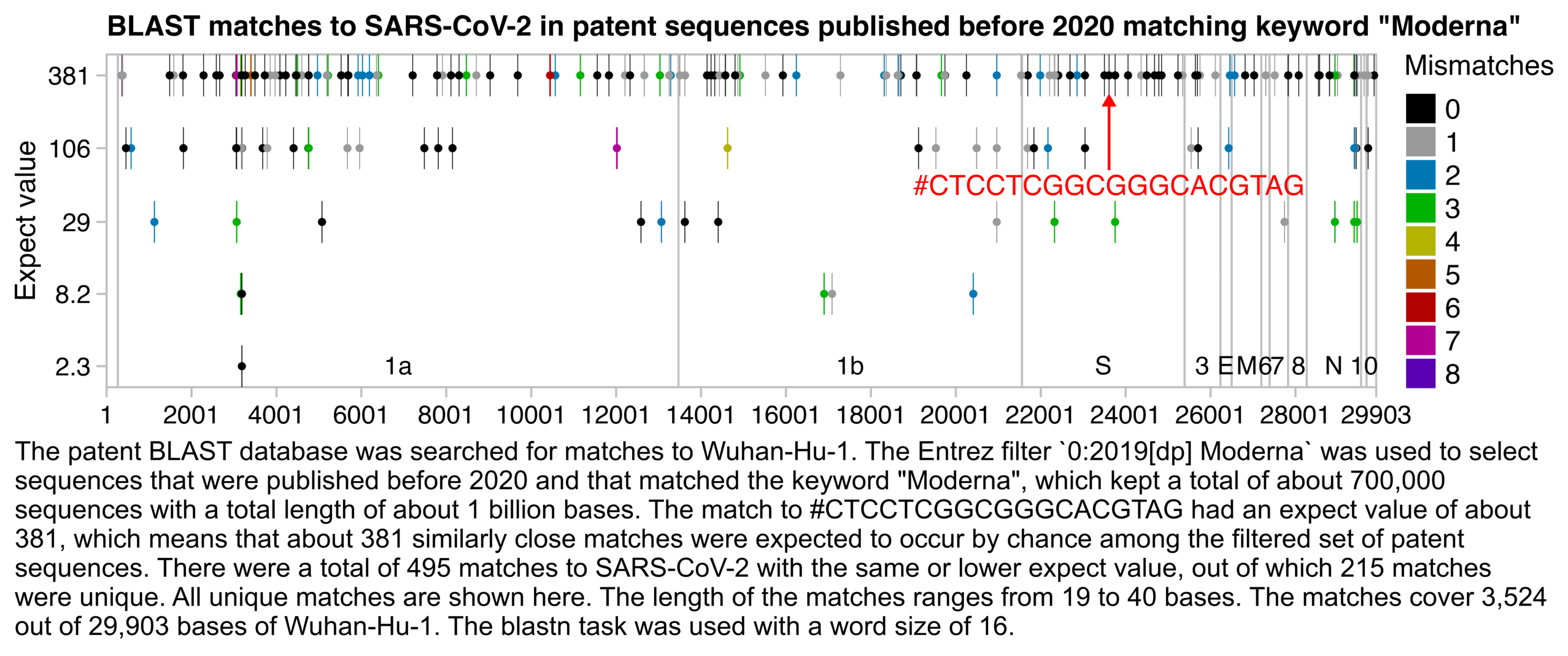
t=fread(" http:// sars2. net/ f/ modernagate- blast. txt") p=unique( t[,.( start=qstart, end=qend, expect=evalue, mismatch=mismatch+ gaps)]) gene=fread( text=" name, start, end 1a, 266, 13468 1b, 13468, 21555 S, 21563, 25384 3, 25393, 26220 E, 26245, 26472 M, 26523, 27191 6, 27202, 27387 7, 27394, 27759 8, 27894, 28259 N, 28274, 29533 10, 29558, 29674") bars=gene[, c( start[ 1], pmean( start[- 1], end[-. N]), end[. N])] exp=. 16; ylim=10^( c(- 1, 1)* exp+ log10( range( p$ expect))) ybreak=unique( p$ expect); xend=29903; xbreak=c( seq( 1, xend, 2e3), xend) color=c( " black", " gray60", hsv( c( 20, 12, 6, 3, 0, 31, 27)/ 36, 1,. 7)) annox=23601+ 10; annoy=10^( log10( 381)- exp); annoy2=10^ mean( log10( ybreak[ 3: 4])) ggplot( p)+ annotate( " rect", xmin=1, xmax=xend, ymin=ylim[ 1], ymax=ylim[ 2], linewidth=. 4, lineend=" square", color=" gray75", fill=NA)+ geom_ vline( xintercept=bars, color=" gray75", linewidth=. 4, lineend=" square")+ geom_ rect( aes( fill=factor( mismatch), xmin=start, xmax=end, ymin=10^( log10( expect)- exp), ymax=10^( log10( expect)+ exp)))+ geom_ point( aes( color=factor( mismatch), x=pmean( start, end), y=expect), size=1. 5, stroke=0)+ geom_ text( data=gene, aes( x=pmean( start, end), y=ylim[ 1], label=name), size=3. 5, hjust=. 5, vjust=-. 7)+ annotate( " segment", x=annox, xend=annox, y=annoy2* 1. 3, yend=annoy, arrow=arrow( type=" closed", length=unit( 4, " pt")), lineend=" butt", linejoin=" mitre", color=" red", size=. 5)+ annotate( " text", x=annox, y=annoy2, size=3. 8, color=" red", label="# CTCCTCGGCGGGCACGTAG")+ labs( x=NULL, y=" Expect value", title=" BLAST matches to SARS- CoV- 2 in patent sequences published before 2020 matching keyword \" Moderna\ " ")+ scale_ x_ continuous( limits=c( 1, xend), breaks=xbreak)+ scale_ y_ log10( breaks=ybreak, labels=ifelse( ybreak> =10, round( ybreak), ybreak))+ scale_ color_ manual( values=color, name=" Mismatches")+ scale_ fill_ manual( values=color, name=" Mismatches")+ coord_ cartesian( clip=" off",
