This downloads FASTA files for the nucleotide and protein sequences of a few SARS-like viruses:
$ printf %s\\n NC_045512\ sars2 NC_004718\ sars1 MN996532\ ratg13 MG772933\ zc45 MG772934\ zxc21 MZ937000\ banal52 >accession $ cat accession|while read l m;do curl -s "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=$l">$m.fa;done $ cat accession|while read l m;do curl -s "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta_cds_aa&id=$l">$m.aa;done $ head -n3 sars2.fa >NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAA CGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTAATAAC $ head -n3 sars2.aa >lcl|NC_045512.2_prot_YP_009724389.1_1 [gene=ORF1ab] [locus_tag=GU280_gp01] [db_xref=GeneID:43740578] [protein=ORF1ab polyprotein] [exception=ribosomal slippage] [protein_id=YP_009724389.1] [location=join(266..13468,13468..21555)] [gbkey=CDS] MESLVPGFNEKTHVQLSLPVLQVRDVLVRGFGDSVEEVLSEARQHLKDGTCGLVEVEKGVLPQLEQPYVF IKRSDARTAPHGHVMVELVAELEGIQYGRSGETLGVLVPHVGEIPVAYRKVLLRKNGNKGAGGHSYGADL
Another option is to use efetch from the NCBI's
E-utilities:
[https://www.ncbi.nlm.nih.gov/books/NBK179288/]
curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh|sh edirect/efetch -db nuccore -id NC_045512 -format fasta edirect/efetch -db nuccore -id NC_045512 -format fasta_cds_aa
The code below creates a single FASTA file for the spike protein sequences of the viruses downloaded in the previous section. It then uses MAFFT to align the sequences and displays the alignment in the human-readable CLUSTAL format. From the output, you can see that the PRRA insert which encodes for the furin cleavage site is missing from each sequence except SARS2, even though BANAL-52 and RaTG13 include the QTNS part of the QTNSPRRA insert:
$ brew install mafft
[...]
$ for x in *.aa;do echo \>${x%%.*};awk '/protein=(spike|surface)/{x=1;next}/^>/{x=0}x' $x;done|mafft --clustalout -
[...]
banal52 GAEHVNNSYECDIPIGAGICASYQTQTNS----RSVASQSIIAYTMSLGAENSVAYSNNS
ratg13 GAEHVNNSYECDIPIGAGICASYQTQTNS----RSVASQSIIAYTMSLGAENSVAYSNNS
sars1 GAEHVDTSYECDIPIGAGICASYHTVSLL----RSTSQKSIVAYTMSLGADSSIAYSNNT
sars2 GAEHVNNSYECDIPIGAGICASYQTQTNSPRRARSVASQSIIAYTMSLGAENSVAYSNNS
zc45 GAEHVNASYECDIPIGAGICASYHTASIL----RSTSQKAIVAYTMSLGAENSIAYANNS
zxc21 GAEHVNASYECDIPIGAGICASYHTASIL----RSTGQKAIVAYTMSLGAENSIAYANNS
*****: ****************:* : **...::*:********:.*:**:**:
[...]
MAFFT is much faster than older multiple sequence aligners like ClustalW. In my benchmark below, aligning the nucleotide sequences of four SARS-like genomes took 5 seconds with MAFFT, 129 seconds with ClustalOmega, 258 seconds with Muscle, and 278 seconds with ClustalW:
brew install mafft muscle clustal-omega clustal-w
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id='{NC_045512,NC_004718.3,MN996532,MG772933}>sars.fa
time mafft --thread 4 sars.fa>/dev/null
time clustalw sars.fa -output=fasta>/dev/null
time clustalo -i sars.fa>/dev/null
time muscle -in sars.fa>/dev/null
First align the nucleotide sequences:
for x in {sars2,sars1,ratg13,zc45,zxc21,banal52}.fa;do echo ">${x%.fa}";sed 1d $x;done|mafft --thread 4 ->all.aln
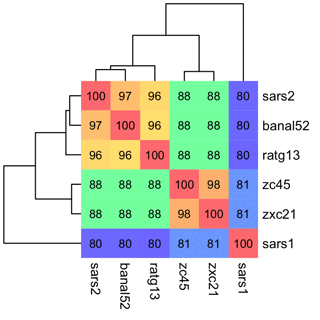
Then run this in R:
install.packages("bio3d")
install.packages("pheatmap")
install.packages("colorspace")
# this uses `bio3d::read.fasta` because the `seqinr` package also has a function called `read.fasta`
t=bio3d::seqidentity(bio3d::read.fasta("all.aln"))
# this reorders the branches of the clustering tree by using the first dimension in an MDS matrix as the weight
hc=as.hclust(reorder(as.dendrogram(hclust(as.dist(1-t))),cmdscale(1-t)[,1]))
# this uses `pheatmap::pheatmap` because the `ComplexHeatmap` package also has a function called `pheatmap`
pheatmap::pheatmap(
100*t,
filename="1.png",
clustering_callback=\(...)hc,
legend=F,
cellwidth=20,
cellheight=20,
fontsize=9,
border_color=NA,
display_numbers=T,
number_format="%.0f",
fontsize_number=8,
number_color="black",
colorRampPalette(colorspace::hex(colorspace::HSV(seq(240,0),.5,1)))(256)
)
Here you can see that SARS2 has about 80% identity with SARS1 and about 97% identity with BANAL-52:
You can find viruses that are similar to SARS2 by going to nucleotide BLAST. [https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn] Enter the accession number of the SARS2 referece genome (NC_045512) to the big text field at the top. Then set the organism to "Coronaviridae (taxid:11118)", click "Add organism", enter "SARS-CoV-2 (taxid:2697049)" and click the "Exclude" checkbox next to it.
When I try to do BLAST searches among all coronaviruses, I usually get an error which says "CPU usage limit was exceeded. You may need to change your search strategy." However I figured out that I can avoid the error by excluding SARS2 from the search results:
Then when you click "BLAST", after a while it should show you the hundred closest matches. Then click "Download" and select "FASTA (complete sequences)" to download all sequences.
You can next repeat the same procedure for other SARS-like sequences, like SARS1 (NC_004718), a Kenyan bat virus BtKY72 (KY352407), a bat virus which is about 4000 nucleotide changes away from SARS1 (DQ648856), a bat virus which is about 100 nucleotide changes away from SARS1 (OK017854), and so on. Then you can concatenate all of the files you downloaded, remove duplicates, exclude partial sequences and monopartite sequences, and add the reference genome of SARS2. And then you can align the sequences with MAFFT:
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=NC_045512'>sars2.fa brew install seqkit cat ~/Downloads/seqdump*.txt sars2.fa|seqkit seq -w 0|paste - -|awk '!a[$1]++'|grep -Ev 'complete cds|partial genome|almost complete genome|genomic sequence, sequence|monopartite'|tr \\t \\n|mafft --thread 4 ->sarslike.fa
In the code above, seqkit -w 0 is used to linearize the
FASTA file so that each sequence is placed on a single long line.
paste - - joins every other line with a tab so that the
title line and sequence are placed on a single line instead of two
lines. awk '!a[$1]++' removes duplicate sequences, even
though you can remove duplicates with seqkit rmdup -s.
If you don't bother creating the FASTA file yourself, you can also download this FASTA file I made: https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5.
The snp-dists utility calculates the number of
nucleotide changes between each pair of sequences in an aligned FASTA
file:
[https://github.com/tseemann/snp-dists]
$ curl https://sars2.net/f/sarbe.aln.xz|xz -dc>sarbe.aln
$ brew install brewsci/bio/snp-dists
[...]
$ snp-dists sarbe.aln>sarbe.dist
[...]
$ head -n4 sarbe.dist|cut -f-4|tr \\t \; # the output of snp-dists only shows sequence IDs but not sequence names
snp-dists 0.7.0;OR233295.1;OR233321.1;OR233320.1
OR233295.1;0;2018;2014
OR233321.1;2018;0;10
OR233320.1;2014;10;0
$ grep ^\> sarbe.aln|cut -c2-|sed $'s/ /\t/;s/, complete genome//'|awk 'NR==FNR{a[$1]=$2;next}FNR==1{for(i=2;i<=NF;i++)$i=$i" "a[$i]}FNR>1{$1=$1" "a[$1]}1' {,O}FS=\\t - sarbe.dist>sarbe.distn
$ head -n4 sarbe.distn|cut -f-4|tr \\t \; # add sequence names to output of snp-dists
snp-dists 0.7.0;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420 surface glycoprotein gene, complete cds;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53 surface glycoprotein gene, complete cds;OR233320.1 Horseshoe bat sarbecovirus isolate Rt22QT48 surface glycoprotein gene, complete cds
OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420 surface glycoprotein gene, complete cds;0;2018;2014
OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53 surface glycoprotein gene, complete cds;2018;0;10
OR233320.1 Horseshoe bat sarbecovirus isolate Rt22QT48 surface glycoprotein gene, complete cds;2014;10;0
$ awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~/Wuhan-Hu-1/)break;next}$1!=x{print$i,$1}' sarbe.distn|sort -n|head
0 NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1
905 MZ937000.1 Bat coronavirus isolate BANAL-20-52/Laos/2020
1138 MN996532.2 Bat coronavirus RaTG13
1159 MZ937001.1 Bat coronavirus isolate BANAL-20-103/Laos/2020
1186 MZ937003.2 Bat coronavirus isolate BANAL-20-236/Laos/2020
1540 MZ081381.1 Betacoronavirus sp. RpYN06 strain bat/Yunnan/RpYN06/2020
1790 MZ937002.1 Bat coronavirus isolate BANAL-20-116/Laos/2020
1804 MZ937004.1 Bat coronavirus isolate BANAL-20-247/Laos/2020
1864 OR233327.1 Horseshoe bat sarbecovirus isolate Rp22DB167 surface glycoprotein gene, complete cds
2087 MW251311.1 Bat coronavirus RacCS264, partial genome
From the output above, you can see that the closest neighbor of SARS2 is BANAL-52 which has 905 nucleotide changes from SARS2.
The default behavior of snp-dists is to only count A, C,
G, and T letters, so it ignores positions where either sequence has a
gap or a degenerate base, and it produces the incorrect result for amino
acid sequences. The -a flag enables counting all letters,
but it also counts positions with a gap so there's no way to ignore
positions with a gap if snp-dists is used with amino acid
sequences.
# `geom_mark_hull` draws the hulls incorrectly in new versions of `ggforce` so you need to install an old version of `ggforce`:
# `install.packages("https://cran.r-project.org/src/contrib/Archive/ggforce/ggforce_0.3.4.tar.gz")`
library(ggplot2)
library(colorspace)
t=read.table("sarslike.dist",sep="\t",row.names=1,header=T)
name=readLines("sarslike.fa")
name=sub("^>","",name[grep("^>",name)])
name=sub(", complete genome","",name)
name=sub(" isolate "," ",name)
name=sub("^([^ ]+) ","",name)
xid="NC_045512.2"
yid="NC_004718.3"
xname=name[match(xid,rownames(t))]
yname=name[match(yid,rownames(t))]
xy=data.frame(x=t[,xid],y=t[,yid])
# # include only samples whose distance to both samples is below 6500 nucleotides
# pick=complete.cases(xy<=5000)
# xy=xy[pick,]
# name=name[pick]
# t=t[pick,pick]
# draw a line between each sample and its two closest neighbors
seg=cbind(xy[rep(1:nrow(xy),each=2),],xy[apply(t,1,\(x)order(x)[2:3]),])
colnames(seg)=paste0("V",1:4)
expand=max(xy)/25
lims=c(-expand,max(apply(xy,2,range))+expand)
k=as.factor(cutree(hclust(as.dist(t)),24))
set.seed(0)
pal=sample(hcl(head(seq(0,360,length.out=nlevels(k)+1),-1),90,60))
ggplot(xy,aes(x,y))+
geom_vline(xintercept=0,color="gray80",size=.3)+
geom_hline(yintercept=0,color="gray80",size=.3)+
geom_abline(linetype="dashed",color="gray80",size=.3)+
geom_segment(data=seg,aes(x=V1,y=V2,xend=V3,yend=V4),color="gray50",size=.1)+
ggforce::geom_mark_hull(aes(color=k,fill=k),concavity=1000,radius=unit(.15,"cm"),expand=unit(.15,"cm"),alpha=.2,size=.15)+
geom_point(aes(color=k),size=.5)+
# geom_text(aes(color=k),label=name,size=2,vjust=-.7)+ # allow overlapping labels
ggrepel::geom_text_repel(aes(color=k),label=name,size=2,max.overlaps=40,segment.size=.2,min.segment.length=.2,box.padding=.15,force=3)+
coord_cartesian(xlim=lims,ylim=lims,expand=F)+
scale_x_continuous(breaks=seq(0,10000,500))+
scale_y_continuous(breaks=seq(0,10000,500))+
labs(x=paste("Distance to",xname),y=paste("Distance to",yname))+
scale_fill_manual(values=pal)+
scale_color_manual(values=pal)+
theme(
axis.text=element_text(size=6),
axis.text.y=element_text(angle=90,vjust=1,hjust=.5),
axis.ticks=element_blank(),
axis.ticks.length=unit(0,"cm"),
axis.title=element_text(size=8),
legend.position="none",
panel.background=element_rect(fill="white"),
panel.border=element_rect(color="gray80",fill=NA,size=.6),
panel.grid.major=element_line(color="gray90",size=.2),
plot.background=element_rect(fill="white")
)
ggsave("1.png",width=10,height=10)
# `geom_mark_hull` draws the hulls incorrectly in new versions of `ggforce` so you need to install an old version of `ggforce`:
# `install.packages("https://cran.r-project.org/src/contrib/Archive/ggforce/ggforce_0.3.4.tar.gz")`
library(ggplot2)
t=read.table("sarslike.dist",sep="\t",row.names=1,header=T)
name=readLines("sarslike.fa")
name=sub("^>","",name[grep("^>",name)])
name=sub(", complete genome","",name)
name=sub(" isolate "," ",name)
name=sub("SARS coronavirus "," ",name)
name=sub("^([^ ]+) ","",name)
# include only samples with 100 or fewer nucleotide changes from the SARS1 reference genome
pick=t[,"NC_004718.3"]<=100
t=t[pick,pick]
name=name[pick]
mds0=cmdscale(t,ncol(t)-1,eig=T)
mds=as.data.frame(mds0$points)
eig=head(mds0$eig,ncol(mds))
pct=paste0("MDS dimension ",1:ncol(mds)," (",sprintf("%.1f",100*eig/sum(eig)),"%)")
k=as.factor(cutree(hclust(as.dist(t)),16))
set.seed(0)
pal=hcl(sample(head(seq(0,360,nlevels(k)+1),-1)),80,70)
seg=cbind(mds[rep(1:nrow(mds),each=2),1:2],mds[apply(t,1,\(x)order(x)[2:3]),1:2])
colnames(seg)=paste0("V",1:4)
ggplot(mds,aes(V1,V2),group=0)+
ggforce::geom_mark_hull(aes(group=k,color=k,fill=k),concavity=1000,radius=unit(.15,"cm"),expand=unit(.15,"cm"),alpha=.2,size=.15)+
geom_segment(data=seg,aes(x=V1,y=V2,xend=V3,yend=V4),color="black",linewidth=.2)+
geom_point(aes(color=k),size=.5)+
# geom_text(aes(color=k),label=name,size=2,vjust=-.7)+
ggrepel::geom_text_repel(aes(color=k),label=name,size=2,max.overlaps=Inf,segment.size=.2,min.segment.length=.2,box.padding=.15,force=3)+
labs(x=pct[1],y=pct[2])+
scale_x_continuous(breaks=seq(-1000,1000,20),expand=expansion(mult=c(.06,.06)))+
scale_y_continuous(breaks=seq(-1000,1000,20),expand=expansion(mult=c(.04,.04)))+
scale_color_manual(values=pal)+
scale_fill_manual(values=pal)+
theme(
axis.ticks=element_line(linewidth=.4,color="gray40"),
axis.ticks.length=unit(-.15,"cm"),
axis.text=element_text(color="black",size=6),
axis.text.x=element_text(margin=margin(.3,0,0,0,"lines")),
axis.text.y=element_text(angle=90,vjust=1,hjust=.5,margin=margin(0,.3,0,0,"lines")),
axis.title=element_text(color="black",size=8),
legend.position="none",
panel.background=element_rect(fill="gray25"),
panel.border=element_rect(color="gray40",fill=NA,linewidth=.6),
panel.grid=element_blank(),
plot.background=element_rect(fill="gray40",color=NA),
plot.title=element_text(size=10,color="black")
)
ggsave("1.png",width=11,height=7)
The red cluster in the middle of the plot is occupied by early human strains of SARS1, and the yellow cluster next to it is occupied by later human stains of SARS2. The bottom left corner of the plot is occupied by lab-created strains like MA5, ExoN1, and the wtic strains, which are all derived from the Urbani strain. Above them there are a couple of infectious clones of the Urbani strain. The bottom right corner of the plot is occupied by samples which were allegedly found in Himalayan palm civets, even though they only have 50-100 nucleotide changes from the reference genome of SARS1:
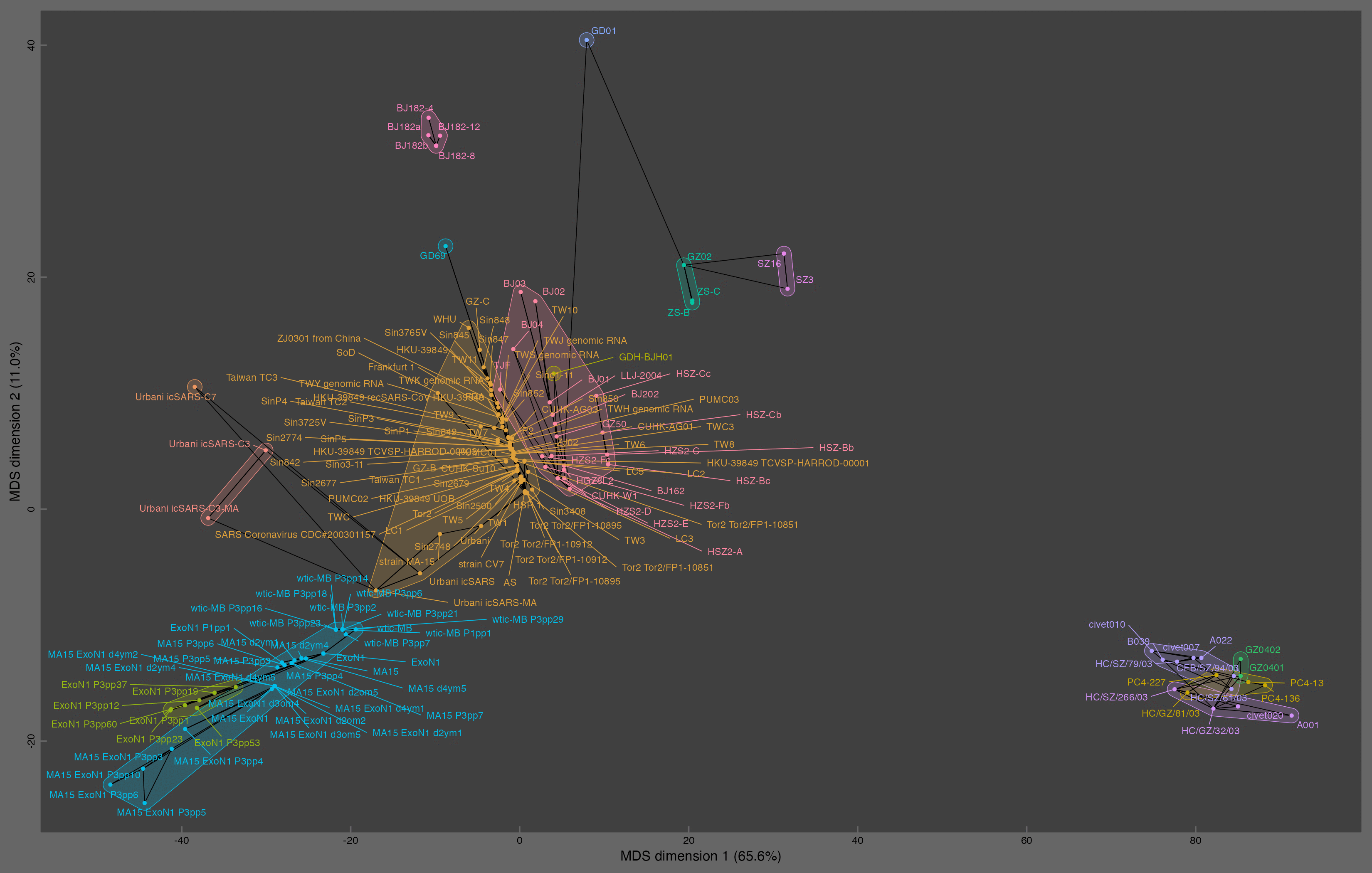
library(circlize)
t=read.table("sarslike.dist",header=T,row.names=1,sep="\t")
name=readLines("sarslike.fa")
name=sub("^>","",name[grep("^>",name)])
name=sub(", complete genome","",name)
name=sub(" isolate "," ",name)
# reorder clustering tree based on value of first dimension in MDS
hc=as.hclust(reorder(as.dendrogram(hclust(as.dist(t),"ward.D2")),cmdscale(t)[,1]))
labels=hc$label[hc$order]
cut=cutree(hc,30)
set.seed(0)
hue=c(0,30,60,90,130,170,210,240,280,320)+15
color=sample(c(hcl(hue,50,50),hcl(hue,40,60),hcl(hue,30,30)))
dend=dendextend::color_branches(as.dendrogram(hc),k=length(unique(cut)),col=color[unique(cut[labels])])
circos.clear()
size=4500
png("1.png",w=size,h=size,res=150)
circos.par(cell.padding=c(0,0,0,0))
circos.initialize(0,xlim=c(0,nrow(t)))
labelheight=.5 # use 50% of height for labels
circos.track(ylim=c(0,1),bg.border=NA,track.height=labelheight,track.margin=c(0,0),panel.fun=\(x,y)
for(i in 1:nrow(t))circos.text(i-.5,0,name[hc$order][i],adj=c(0,.5),facing="clockwise",niceFacing=T,cex=.9,col=color[cut[labels[i]]]))
circos.track(ylim=c(0,attr(dend,"height")),track.height=1-labelheight-.01,track.margin=c(0,.005),bg.border=NA,panel.fun=\(x,y)circos.dendrogram(dend))
circos.clear()
dev.off()
# remove empty space around the plot but add back a 24px margin
system("mogrify -gravity center -trim -border 24 -bordercolor white 1.png")
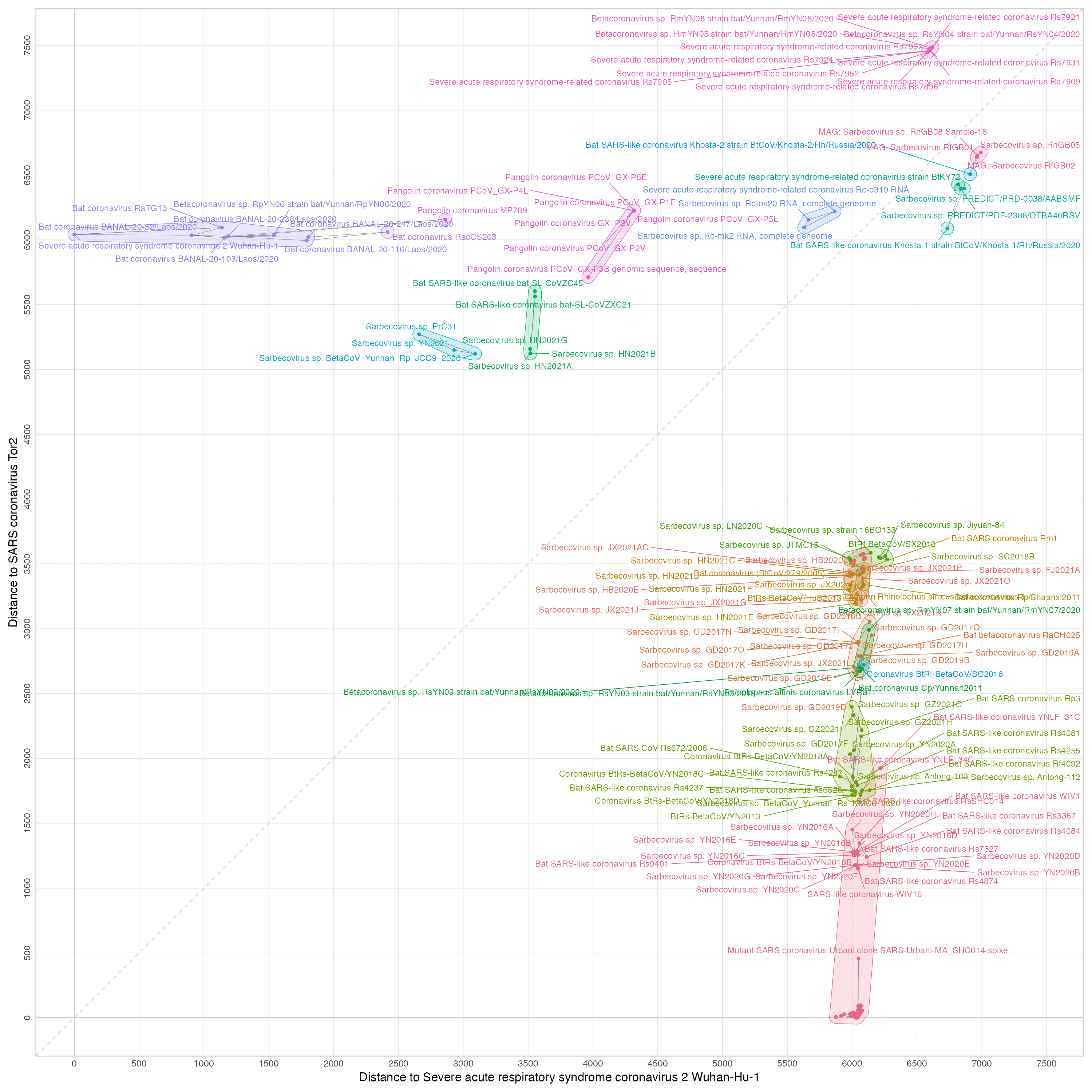
This excludes genomes whose distance to either SARS1 or SARS2 is below 500:
t=read.table("sarslike.dist",header=T,row.names=1,sep="\t")
name=readLines("sarslike.fa")
name=sub("^>","",name[grep("^>",name)])
name=sub(", complete genome","",name)
name=sub(" isolate "," ",name)
colnames(t)=rownames(t)=name
s1="NC_004718.3 SARS coronavirus Tor2"
s2="NC_045512.2 Severe acute respiratory syndrome coronavirus 2 Wuhan-Hu-1"
dist=data.frame(sum=t[,s1]+t[,s2],d1=t[,s1],d2=t[,s2],row.names=rownames(t))
dist=dist[rowSums(dist[,-1]<=500)==0,]
# format table with row names on the right side
fotar=\(x)writeLines(apply(cbind(apply(rbind(colnames(x),x),2,\(c)sprintf(paste0("%",max(nchar(c)),"s"),c)),c("",rownames(x))),1,paste,collapse=" "))
fotar(head(dist[order(dist[,1]),],20))
Output:
sum d1 d2 6940 6035 905 MZ937000.1 Bat coronavirus BANAL-20-52/Laos/2020 7176 6017 1159 MZ937001.1 Bat coronavirus BANAL-20-103/Laos/2020 7193 1150 6043 KT444582.1 SARS-like coronavirus WIV16 7196 1176 6020 OK017856.1 Sarbecovirus sp. YN2020F 7199 1153 6046 KY417150.1 Bat SARS-like coronavirus Rs4874 7201 1177 6024 OK017857.1 Sarbecovirus sp. YN2020G 7211 1175 6036 OK017852.1 Sarbecovirus sp. YN2020B 7213 1176 6037 OK017855.1 Sarbecovirus sp. YN2020E 7213 1176 6037 OK017854.1 Sarbecovirus sp. YN2020D 7213 6027 1186 MZ937003.2 Bat coronavirus BANAL-20-236/Laos/2020 7214 1177 6037 OK017853.1 Sarbecovirus sp. YN2020C 7232 6094 1138 MN996532.2 Bat coronavirus RaTG13 7264 1254 6010 MK211376.1 Coronavirus BtRs-BetaCoV/YN2018B 7277 1273 6004 OK017849.1 Sarbecovirus sp. YN2016C 7278 1255 6023 KY417152.1 Bat SARS-like coronavirus Rs9401 7292 1283 6009 OK017848.1 Sarbecovirus sp. YN2016B 7294 1280 6014 OK017851.1 Sarbecovirus sp. YN2016E 7294 1279 6015 OK017850.1 Sarbecovirus sp. YN2016D 7299 1283 6016 OK017847.1 Sarbecovirus sp. YN2016A 7300 1256 6044 KY417151.1 Bat SARS-like coronavirus Rs7327
$ mkdir xml
$ awk -F'[> ]' '/^>/{print$2}' sarslike.fa|head -n4|while read l;do curl -s "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&retmode=xml&id=$l"|xml fo -D>xml/$l;done
$ brew install xmlstarlet
[...]
$ awk -F'[> ]' '/^>/{print$2}' sarslike.fa|head -n4|while read x;do printf %s\\n "$(xml sel -t -v //GBSeq_primary-accession xml/$x)" "$(xml sel -t -v //GBSeq_definition xml/$x)" "$(gdate -d$(xml sel -t -v //GBSeq_create-date xml/$x) +%F)" "$(xml sel -t -v //GBReference_title xml/$x|tail -n1)" "$(xml sel -t -v '//GBQualifier[GBQualifier_name="collection_date"]/GBQualifier_value' xml/$x)" "$(xml sel -t -v '//GBQualifier[GBQualifier_name="country"]/GBQualifier_value' xml/$x)"|tr \; _|paste -sd\; -;done
FJ882959;SARS coronavirus MA15 ExoN1 isolate P3pp6, complete genome;2009-12-13;A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice;26-Nov-2008;USA: Tennessee
FJ882951;SARS coronavirus MA15 ExoN1 isolate P3pp3, complete genome;2009-12-13;A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice;26-Nov-2008;USA: Tennessee
FJ882962;SARS coronavirus MA15 ExoN1 isolate P3pp10, complete genome;2009-12-13;A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice;18-Dec-2008;USA: Tennessee
FJ882942;SARS coronavirus MA15 ExoN1 isolate P3pp5, complete genome;2009-12-13;A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice;26-Nov-2008;USA: Tennessee
When I searched for the nucleotide sequence that encodes for the PRRA furin cleavage site in SARS2, it was not included in any other genome:
$ curl -Lso sarslike.fa 'https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5' $ sed 's/, complete genome//' sarslike.fa|tr \ _|seqkit locate -p cctcggcgggca|column -ts$'\t' seqID patternName pattern strand start end matched NC_045512.2_Severe_acute_respiratory_syndrome_coronavirus_2_isolate_Wuhan-Hu-1 cggcgggcacgt cggcgggcacgt + 24286 24297 cggcgggcacgt $ seqkit translate<<<$'>a\ncctcggcgggca'|sed 1d PRRA
A Chinese paper from 2021 found that influenza samples collected in France between 2007 and 2012 were contaminated with SARS1. [https://www.sciencedirect.com/science/article/pii/S2590053621001075] The SARS1 contaminants matched the lab-created wtic-MB and ExoN1 strains of SARS1. Kevin McKernan posted instructions on how you can assemble the SARS1 sequences in the French influenza samples. [https://x.com/Kevin_McKernan/status/1472265147221786639] I expanded his instructions to also generate the consensus sequence:
brew install sratoolkit bwa samtools bcftools cutadapt # cutadapt is needed by trim_galore
vdb-config # configure sra-tools in order to use fasterq-dump
wget https://github.com/FelixKrueger/TrimGalore/archive/0.6.10.tar.gz;tar -xf TrimGalore-0.6.10.tar.gz;cp TrimGalore-0.6.10/trim_galore /usr/local/bin
x=ERR1091914;fasterq-dump $x # download the raw reads of one sample (the other two samples were ERR1091919 and ERR1091920)
trim_galore $x.fastq
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=NC_004718.3&rettype=fasta'>sars1.fa
bwa index sars1.fa
bwa mem -t 4 sars1.fa ${x}_trimmed.fq|samtools view -Sb ->$x.bam
samtools sort $x.bam -o $x.sort.bam
samtools index $x.sort.bam
bcftools mpileup -Ou -f sars1.fa $x.sort.bam|bcftools call -mv -Oz -o calls.vcf.gz
bcftools index calls.vcf.gz
<sars1.fa bcftools consensus calls.vcf.gz>$x.fa
The code below downloads the raw reads from the Wu et al. 2020 paper where the reference genome of SARS2 was published. [https://www.nature.com/articles/s41586-020-2008-3] It then uses MEGAHIT to perform de-novo assembly:
brew install megahit -s # -s compiles from source
brew install trimmomatic sratoolkit
vdb-config # configure sra-tools in order to use fasterq-dump
fasterq-dump SRR10971381 # download raw reads from the NCBI's sequence read archive (about 2 GB)
trimmomatic PE SRR10971381_{1,2}.fastq {1,2}{,un}paired.fastq.gz AVGQUAL:20 HEADCROP:12 LEADING:3 TRAILING:3 MINLEN:75 -threads 4
megahit -1 1paired.fastq.gz -2 2paired.fastq.gz -o out
# megahit -1 SRR10971381_1.fastq -2 SRR10971381_2.fastq -o out2 # try running MEGAHIT with no trimming
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=NC_045512'>sars2.fa
awk 'length>max{max=length;o=$0}END{print o}' out/final.contigs.fa|rev|sed y/ATCG/TAGC/|sed 1i\>longest_contig|cat - sars2.fa|mafft --clustalout -
The last line above prints the reverse complement of the longest
contiguous sequence and aligns it to the SARS2 reference genome.
Sometimes the longest contig is the reverse complement of the genome and
sometimes not, so you have to check both versions. You can get the
reverse complement with rev|sed y/ATCG/TAGC/ or
seqtk seq -rp.
The parameters I used with Trimmomatic are probably not optimal, but I just copied them from this article: https://research.csc.fi/metagenomics_quality.
I used brew install megahit -s to compile MEGAHIT from
source, because without the -s option I got the error
dyld: Library not loaded: /usr/local/opt/gcc/lib/gcc/9/libgomp.1.dylib
(because I had GCC 11 and not 9).
When I didn't use Trimmomatic, my longest contiguous sequence was 29802 bases long and it was missing the last 102 bases of the third version of Wuhan-Hu-1 and it had one extra base at the beginning. When I did use Trimmomatic, my longest contiguous sequence was now 29875 bases long, it was only missing the last 30 bases from Wuhan-Hu-1, but it still had one extra base at the beginning. I probably used the wrong parameters with Trimmomatic though, or I'm still missing some steps in the pipeline.
When I ran MEGAHIT without Trimmomatic, my second-longest contig was 16,037 bases long and its best match at BLAST was "Leptotrichia hongkongensis JMUB5056 DNA, complete genome" with 99.18% identity and 99% query coverage. My third-longest contig was 13,657 bases long and its best match at BLAST was "Select seq LR778174.1 Veillonella parvula strain SKV38 genome assembly, chromosome: 1" with 99.82% identity. My fourth-longest contig was 11,777 bases long and its best match was "Leptotrichia sp. oral taxon 212 strain W10393, complete genome" with 99.24% identity. I didn't find any matches for my fifth-longest contig, but my sixth-longest contig was 8,062 bases long and its best match was "Select seq CP068263.2 Homo sapiens isolate CHM13 chromosome 15" with 99.89% identity. From the "Analysis" tab of NCBI's sequence read archive, you can see which organisms the reads are estimated to come from. [https://trace.ncbi.nlm.nih.gov/Traces/?view=run_browser&acc=SRR10971381&display=analysis] For SRR10971381, 61% of reads are unidentified, 33% are classified under bacteria, 5% under eukaryotes, and only 0.2% under viruses. So the ratio of 33% bacteria to 5% eukaryotes is consistent with my BLAST searches, where three out of four reads were from bacteria and the fourth was a human read.
Limeng Yan has pointed out that in the Zhoushan bat viruses ZC45 and ZXC21, the envolope protein is identical to the reference genome of SARS2. In the second Yan report, she pointed out that the envelope protein was also identical in many of the supposed pangolin and bat viruses that are similar to SARS2 and that were published in 2020 and 2021: [https://zenodo.org/record/4650821]

The code below takes the file of nucleotide sequences I've used as an example in this thread, downloads amino acid sequences for each virus, and prints the names of viruses with an identical envelope protein to Wuhan-Hu-1 along with the number of nucleotide changes from Wuhan-Hu-1:
$ curl -Lso sarslike.fa 'https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5'
$ brew install brewsci/bio/snp-dists
$ snp-dists sarslike.fa>sarslike.dist
$ sed -n 's/^>//p' sarslike.fa|cut -d\ -f1|while read l;do curl -s "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta_cds_aa&id=$l";done>sarslike.aa
$ 1li()(seqkit seq -w 0 "$@"|paste - -) # convert a FASTA file to a format with one entry per line
$ masot()(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i==x)break;next}$1!=x{print$i,$1}' x="$1" "${2--}"|sort -n) # print a column which matches the specified column name from a matrix and display the sorted values with row names
$ 1li<sarslike.aa|grep $'\t'$(1li<sarslike.aa|grep 'NC_045512.*envelope'|cut -f2)\$|sed 's/_prot_.*//;s/>lcl|//'|awk 'NR==FNR{a[$0];next}$2 in a' - <(masot NC_045512.2 sarslike.dist)|awk 'NR==FNR{a[$1]=$0;next}{print$1,a[$2]}' <(sed -n 's/^>//p' sarslike.fa) -|sed 's/, complete genome//'
905 MZ937000.1 Bat coronavirus isolate BANAL-20-52/Laos/2020
1138 MN996532.2 Bat coronavirus RaTG13
1186 MZ937003.2 Bat coronavirus isolate BANAL-20-236/Laos/2020
1540 MZ081381.1 Betacoronavirus sp. RpYN06 strain bat/Yunnan/RpYN06/2020
2860 MT121216.1 Pangolin coronavirus isolate MP789
3092 OK287355.1 Sarbecovirus sp. isolate BetaCoV_Yunnan_Rp_JCC9_2020
3518 OK017803.1 Sarbecovirus sp. isolate HN2021A
3518 OK017804.1 Sarbecovirus sp. isolate HN2021B
3553 MG772933.1 Bat SARS-like coronavirus isolate bat-SL-CoVZC45
3554 MG772934.1 Bat SARS-like coronavirus isolate bat-SL-CoVZXC21
4312 MT040335.1 Pangolin coronavirus isolate PCoV_GX-P5L
4312 MT072864.1 Pangolin coronavirus isolate PCoV_GX-P2V
4313 MW532698.1 Pangolin coronavirus isolate GX_P2V
4314 MT040333.1 Pangolin coronavirus isolate PCoV_GX-P4L
4317 MT040334.1 Pangolin coronavirus isolate PCoV_GX-P1E
4319 MT040336.1 Pangolin coronavirus isolate PCoV_GX-P5E
5629 LC663959.1 Sarbecovirus sp. Rc-mk2 RNA, complete geneome
Another option is to use seqkit locate, even though it
will also match proteins that have additional insertions at the
beginning or end of the sequence:
seqkit grep -nrp 'NC_045512.*envelope' sarslike.aa|seqkit locate -f- sarslike.aa.
However maybe there's nothing that unusual about the envelope protein being idential, because there's viruses that have over 3000 nucleotide changes from SARS1 but that still have an identical envelope protein to SARS1:
$ 1li<sarslike.aa|grep $'\t'$(1li<sarslike.aa|grep 'NC_004718.*envelope'|cut -f2)\$|sed 's/_prot_.*//;s/>lcl|//'|awk 'NR==FNR{a[$0];next}$2 in a' - <(masot NC_004718.3 sarslike.dist)|awk 'NR==FNR{a[$1]=$0;next}{print$1,a[$2]}' <(sed -n 's/^>//p' sarslike.fa) -|sed 's/, complete genome//'|tail -n1
3549 GQ153548.1 Bat SARS coronavirus HKU3-13
In order to return similar proteins in addition to identical proteins, you can use BLAST:
$ brew install blast
[...]
$ makeblastdb -in sarslike.aa -dbtype prot
[...]
$ blastp -query <(seqkit grep -nrp 'NC_004718.*envelope' sarslike.aa) -db sarslike.aa
[...]
$ seqkit grep -nrp NC_004718.\*envelope sarslike.aa|blastp -db sarslike.aa -outfmt 6|head -n2|tr \\t \; # `-outfmt 6` uses TSV output
lcl|NC_004718.3_prot_YP_009825054.1_6;lcl|MT308984.1_prot_QJE50592.1_6;100.000;76;0;0;1;76;1;76;7.96e-49;146
lcl|NC_004718.3_prot_YP_009825054.1_6;lcl|GU553363.1_prot_ADC35486.1_6;100.000;76;0;0;1;76;1;76;7.96e-49;146
$ seqkit grep -nrp NC_004718.\*envelope sarslike.aa|blastp -db sarslike.aa -outfmt 6|cut -f2,5|sed $'s/lcl|//;s/_prot_[^\t]*//'|awk 'NR==FNR{a[$1]=$2;next}$2 in a{print a[$2],$0}' - <(masot NC_004718.3 sarslike.dist)|awk 'NR==FNR{a[$1]=$0;next}{print$1,$2,a[$3]}' <(sed -n s/^\>//p sarslike.fa) -|sed 's/, complete genome//'|sort -nk1,1 -k2,2|head -n2
0 0 JX163927.1 SARS coronavirus Tor2 isolate Tor2/FP1-10851
0 1 JX163923.1 SARS coronavirus Tor2 isolate Tor2/FP1-10912
In the output of the last command above, the first column shows the number of amino acid changes in the envelope protein and the second column shows the number of nucleotide changes in the whole genome.
To calculate a distance matrix for the number of nucleotide or amino
acid changes in an aligned FASTA file, you can use Biostrings:
stringDist(readDNAStringSet("input.fa"),"hamming")). The
following code finds pairs of viruses where there's two or fewer amino
acid changes in the spike protein, and it then sorts the results by the
number of nucleotide changes in the whole genome:
# install.packages("BiocManager")
# BiocManager::install("Biostrings")
library(Biostrings)
d1=as.matrix(stringDist(readDNAStringSet("sarslike.fa"),"hamming"))
name=sub(", complete genome","",rownames(d1))
name=sub(" isolate "," ",name)
id=sub(" .*","",rownames(d1))
rownames(d1)=colnames(d1)=id
name=setNames(name,id)
d2=as.matrix(stringDist(readAAStringSet("spike.aln"),"hamming"))
colnames(d2)=rownames(d2)=sub("_prot_.*","",sub("lcl\\|","",rownames(d2)))
d1=d1[rownames(d2),rownames(d2)]
long=data.frame(c(d1),c(d2),name[rownames(d1)[row(d1)]],name[colnames(d1)[col(d1)]])
long=long[long[,3]<long[,4],]
pick=long[long[,2]<=2,]
pick=pick[rev(tail(order(pick[,1]),10)),]
write.table(pick,sep=";",quote=F,row.names=F,col.names=F)
Output:
1044;0;KC881005.1 Bat SARS-like coronavirus RsSHC014;MT308984.1 Mutant SARS coronavirus Urbani clone SARS-Urbani-MA_SHC014-spike 871;1;KY417144.1 Bat SARS-like coronavirus Rs4084;MT308984.1 Mutant SARS coronavirus Urbani clone SARS-Urbani-MA_SHC014-spike 786;1;KY417151.1 Bat SARS-like coronavirus Rs7327;KY417152.1 Bat SARS-like coronavirus Rs9401 559;2;KC881006.1 Bat SARS-like coronavirus Rs3367;KF367457.1 Bat SARS-like coronavirus WIV1 432;0;OL674074.1 Severe acute respiratory syndrome-related coronavirus Rs7896;OL674078.1 Severe acute respiratory syndrome-related coronavirus Rs7921 425;0;OK017844.1 Sarbecovirus sp. JX2021O;OK017845.1 Sarbecovirus sp. JX2021P 421;0;OL674074.1 Severe acute respiratory syndrome-related coronavirus Rs7896;OL674077.1 Severe acute respiratory syndrome-related coronavirus Ra7909 417;0;OL674074.1 Severe acute respiratory syndrome-related coronavirus Rs7896;OL674080.1 Severe acute respiratory syndrome-related coronavirus Rs7931 409;1;KC881005.1 Bat SARS-like coronavirus RsSHC014;KY417144.1 Bat SARS-like coronavirus Rs4084 397;0;OL674074.1 Severe acute respiratory syndrome-related coronavirus Rs7896;OL674076.1 Severe acute respiratory syndrome-related coronavirus Rs7907
From the first line of the output above, you can see that the viruses titled "Bat SARS-like coronavirus RsSHC014" and "Mutant SARS coronavirus Urbani clone SARS-Urbani-MA_SHC014-spike" have an identical spike protein even though they have 1044 nucleotide changes in the whole genome. The mutant virus was published in a paper from 2015 by the Wuhan Institute of Virology whose last author was Ralph Baric. [https://www.ncbi.nlm.nih.gov/nuccore/MT308984] They used a mouse-adapted strain of SARS1 called MA15 as the backbone and inserted the spike protein from the SHC014 virus, which had been published in a paper from 2013 whose last author was Zhengli Shi and whose authors also included Peter Daszak. The mutant virus was also mentioned in a report which the EcoHealth Alliance produced in order to renew their research grant from the NIH: "In collaboration with Ralph Baric (UNC), we used the SARS-CoV reverse genetics system to generate a chimeric virus with a mouse-adapted SARS-CoV backbone expressing SHC014 S protein with 10% sequence divergence from SARS-CoV S. This chimera replicated in primary human airway epithelium, using the human ACE2 receptor to enter into cells." [https://s3.documentcloud.org/documents/21055989/understanding-risk-bat-coronavirus-emergence-grant-notice.pdf]
From the second line of the output above, you can see that the spike protein of Rs4084 has only one amino acid change from the MA15-SHC014 mutant. Rs4084 was published in a paper by the Wuhan Institute of Virology from 2017 whose last author was Zhengli Shi. [https://www.ncbi.nlm.nih.gov/nuccore/KY417144.1]
You can also use awk to calculate the distance matrix for the number of amino acid changes in the spike protein:
sed 's/>lcl|\(.*\)_prot_.*/>\1/' spike.aln|awk '{printf(/^>/?"\n%s\t":"%s"),$0}'|grep .|paste - -|cut -c2-|gawk -F\\t '{n[NR]=$1;split($2,a,"");l=length(a);for(i=1;i<=l;i++)x[NR][i]=a[i]}END{for(i=1;i<=NR;i++)printf"\t%s",n[i];print"";for(i=1;i<=NR;i++){printf"%s",n[i];for(j=1;j<=NR;j++){s=0;for(k=1;k<=l;k++)if(x[i][k]!=x[j][k])s++;printf"\t%s",s};print""}}'>spike.dist
# install.packages("BiocManager")
# BiocManager::install("ggtree")
# BiocManager::install("Biostrings")
library(ggtree)
library(Biostrings)
t=as.matrix(stringDist(readDNAStringSet("https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5"),"hamming"))
name=rownames(t)
name=sub(", complete genome","",name)
name=sub(" isolate "," ",name)
name=sub(", genomic sequence, sequence"," ",name)
rownames(t)=name
pick=t[grep("Wuhan-Hu-1",rownames(t)),]<=5000
t=t[pick,pick]
set.seed(0)
hue=c(0,30,60,90,130,170,210,240,280,320)+15
color=c("black",sample(c(hcl(hue,50,30),hcl(hue,60,60))))
# reorder branches based on first dimension in MDS
hc=as.hclust(reorder(as.dendrogram(hclust(as.dist(t))),cmdscale(t)[,1]))
cut=cutree(hc,20)
p=ggtree(hc,size=.15,ladderize=F) # `ladderize=F` is needed to preserve the order of the tree
clades=sapply(split(names(cut),cut),\(n)MRCA(p,n))
p=groupClade(p,clades,group_name="subtree")+aes(color=subtree)
p%<+%data.frame(label=names(cut))+
geom_tiplab(size=2)+
scale_color_manual(values=color)+
xlim(0,1.3*max(hc$height))+ # control ratio of label width to tree width
theme(legend.position="none")
ggsave("1.png",height=.09*nrow(t),width=6)
In the output below, the first date on each line is the date when the sequence was published at GenBank and the second date is the collection date of the sample. There are only two viruses that have less than 5000 nucleotide changes from Wuhan-Hu-1 and that had been published at GenBank before 2020. They are the Zhoushan bat viruses ZX45 and ZXC21, which were submitted to GenBank by "Institute of Military Medicine Nanjing Command":

In a pre-print published on February 2th 2020, a program called FAST-NA was used to identify unique regions within the genome of SARS2 by finding k-mers of amino acid sequences which did not appear in other coronaviruses : [https://www.biorxiv.org/content/10.1101/2020.01.31.929497v1.full]
To identify unique sequences, we adapted FAST-NA, a software tool for screening DNA synthesis orders for pathogens(2,3) that uses methods for automatic signature generation developed originally for cybersecurity malware detection(4). In particular, FAST-NA compares all k-mer sequences of a collection of target sequences to a collection of contrasting sequences in order to identify all k-mer sequences that are unique to the target population. These unique sequences are diagnostic of membership in the population, whereas shared sequences indicate structure that is conserved to some degree.
Here, we applied FAST-NA to identify all of the unique 10-mer sequences in all of the amino acid sequences for 2019-nCoV then available from NCBI: 63 amino acid sequences available in NCBI, comprising a total of 49379 amino acids (5-8). For contrasting sequences, we used a July, 2019 snapshot of all protein sequences in family Coronaviridae available from NCBI, a total of 50574 sequences comprising a total of approximately 40 million residues. The resulting collection of unique 10-mer amino acids sequences were then concatenated where overlapping within the same parent sequence and trimmed to remove non-unique flanking portions.
The FAST-NA program is not available for free, so I wrote my own redneck version of the program. My dataset was about an order of magnitude smaller because it included only SARS-like viruses instead of all coronaviruses, even though I downloaded all viruses available in 2023 and not 2019 so my dataset also includes sequences like RaTG13 and BANAL-52:
$ curl -Lso sarslike.fa 'https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5'
$ sed -n 's/^>//p' sarslike.fa|cut -d\ -f1|while read l;do curl -s "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta_cds_aa&id=$l";done>sarslike.aa
$ brew install seqkit
[...]
$ seqkit seq -w0 sarslike.aa|paste - -|grep NC_045512|sed $'s/.*gene=//;s/\][^\t]*//'|grep -v ORF1a\ |awk '{l=length($2);for(i=1;i<=l-k+1;i++)print$1,i,substr($2,i,k)}' k=10 >kmer # ORF1a is omitted because it is also included in ORF1ab
$ awk '{print">"NR"\n"$3}' kmer|seqkit locate -Ff - <(seqkit grep -vnrp NC_045512 sarslike.aa)>match # `-F` uses FM-index which makes searching for a long list of patterns faster
$ awk 'NR==FNR{a[$7];next}!($3 in a)' match kmer|head -n4 # show unique 10-mers along with their protein and starting position within the protein
ORF1ab 29 RGFGDSVEEV
ORF1ab 30 GFGDSVEEVL
ORF1ab 31 FGDSVEEVLS
ORF1ab 32 GDSVEEVLSE
$ awk 'NR==FNR{a[$7];next}!($1 in b){b[$1]=0}!($3 in a){b[$1]++}END{for(i in b)print i,b[i]}' match kmer|awk 'NR==FNR{a[$1]++;next}{print$1,$2/a[$1],$2,a[$1]}' OFMT=%.4f kmer -|sort -rnk2|column -t # show the ratio of unique k-mers to total k-mers within each protein
ORF8 0.2679 30 112
ORF3a 0.1053 28 266
S 0.0791 100 1264
ORF7b 0.0588 2 34
ORF1ab 0.0230 163 7087
M 0.0141 3 213
N 0.0098 4 410
ORF7a 0 0 112
ORF6 0 0 52
ORF10 0 0 29
E 0 0 66
$ awk 'NR==FNR{a[$7];next}!($3 in a)' match kmer|awk 'NR==1||$1!=prev1||lastprint==NR-1{start=$2;prev1=$1;prev2=$2;next}$2!=prev2+1{print prev1,start,prev2+k-1;lastprint=NR;prev1=$1;prev2=$2;next}{prev1=$1;prev2=$2}END{if(lastprint!=NR)print $1,start,$2+k-1}' k=10|awk '{print$0,$3-$2+1}' # show the ranges within each protein which feature unique k-mers
ORF1ab 29 47 19
ORF1ab 272 289 18
ORF1ab 368 385 18
ORF1ab 856 868 13
ORF1ab 981 991 11
ORF1ab 993 1008 16
ORF1ab 1008 1017 10
ORF1ab 1152 1168 17
ORF1ab 1385 1402 18
ORF1ab 1726 1742 17
ORF1ab 1771 1788 18
ORF1ab 1787 1804 18
ORF1ab 1814 1831 18
ORF1ab 2025 2038 14
ORF1ab 2074 2091 18
ORF1ab 2397 2410 14
ORF1ab 3135 3152 18
ORF1ab 3598 3615 18
S 23 41 19
S 42 59 18
S 68 85 18
S 210 227 18
S 338 355 18
S 364 379 16
S 395 411 17
S 490 507 18
S 511 528 18
ORF3a 1 19 19
ORF3a 20 37 18
ORF8 1 19 19
ORF8 18 35 18
N 28 40 13
In a paper from 2020 by Ren et al. titled "Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study", they described de-novo assembly of a sample of SARS2 collected in December 2019. [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7147275/] Compared to the third version of Wuhan-Hu-1 at GenBank, the EPI_ISL_402123/IPBCAMS-WH-01 sequence from the Ren et al. paper is missing the last 4 bases of the poly(A) tail and it has 3 nucleotide changes in the region of the ORF1a protein:
$ curl https://download.cncb.ac.cn/gwh/Viruses/Severe_acute_respiratory_syndrome_coronavirus_2_IPBCAMS-WH-01_GWHABKF00000000/GWHABKF00000000.genome.fasta.gz|gzip -dc>ren
$ curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947'>wuhanhu1
$ cat ren wuhanhu1|mafft --quiet -|awk '{printf(/^>/?"\n%s\n":"%s"),$0}'|grep .>temp.aln;paste <(sed -n 2p temp.aln|grep -o .) <(sed -n 4p temp.aln|grep -o .)|awk '$1!=$2{print NR,$1,$2}'
3778 g a
8388 g a
8987 a t
29900 - a
29901 - a
29902 - a
29903 - a
The shell code above only works with pairwise alignments, but the following code can also be used to compare three or more sequences:
> m=t(as.matrix(Biostrings::readDNAStringSet("temp.aln")))
> pick=which(rowSums(m!=m[,1])>0)
> write.table(`rownames<-`(m[pick,],pick),sep=";",quote=F,col.names=NA) # without `col.names=NA` the semicolon at the start of the header line is omitted
;GWHABKF00000001 OriSeqID=IPBCAMS-WH-01_2019 Len=29899;MN908947.3 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
3778;G;A
8388;G;A
8987;A;T
29900;-;A
29901;-;A
29902;-;A
29903;-;A
Or the same in gawk (this doesn't work with
nawk which doesn't support arrays of arrays):
$ awk 'NR>1{gsub("\t"," ");sub("\n","\t");gsub("\n","");print}' RS=\> temp.aln|awk -F\\t '{o=o";"$1;l=length($2);for(i=1;i<=l;i++){x=substr($2,i,1);a[i][NR]=x;b[i][x]}}END{print o;for(i in b)if(length(b[i])>1){o=i;for(j=1;j<=NR;j++)o=o";"a[i][j];print o}}'
;GWHABKF00000001 OriSeqID=IPBCAMS-WH-01_2019 Len=29899;MN908947.3 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
3778;g;a
8388;g;a
8987;a;t
29900;-;a
29901;-;a
29902;-;a
29903;-;a
In February 2020 the WHO published a PDF which featured tables of primer and probe sequences used in seven different RT-PCR protocols. [https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf] I tried searching for the first primer pair mentioned in the PDF to see if it matches other viruses besides SARS2, but it only matched RaTG13:
$ seqkit amplicon -F atgagcttagtcctgttg -R ctccctttgttgtgttgt sarslike.fa # `-F` specifies forward primer and `-R` specifies reverse primer
[INFO] 1 primer pair loaded
>MN996532.2 Bat coronavirus RaTG13, complete genome
atgagcttagtcctgttgcactacgacagatgtcttgtgctgccggtactacacaaactg
cttgcactgatgacaatgcgttagcttactacaacacaacaaagggag
>NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
atgagcttagtcctgttgcactacgacagatgtcttgtgctgccggtactacacaaactg
cttgcactgatgacaatgcgttagcttactacaacacaacaaagggag
$ awk '{printf(/^>/?"\n%s\n":"%s"),$0}' sarslike.fa|grep .|paste - -|cut -c2-|awk '{if(match($2,x))print RSTART,RLENGTH,substr($2,RSTART,RLENGTH),$1}' {,O}FS=\\t x=atgagcttagtcctgttg.\*$(rev<<<ctccctttgttgtgttgt|sed y/atcg/tagc/)
13118 108 atgagcttagtcctgttgcactacgacagatgtcttgtgctgccggtactacacaaactgcttgcactgatgacaatgcgttagcttactacaacacaacaaagggag MN996532.2 Bat coronavirus RaTG13, complete genome
13118 108 atgagcttagtcctgttgcactacgacagatgtcttgtgctgccggtactacacaaactgcttgcactgatgacaatgcgttagcttactacaacacaacaaagggag NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
$ seqkit translate<<<$'>a\ncctcggcgggca'|sed 1d
PRRA
$ R -e 'if(!require("BiocManager"))install.packages("BiocManager");BiocManager::install("Biostrings")'
[...]
$ Rscript -e 'writeLines(as.character(Biostrings::translate(Biostrings::DNAString("cctcggcgggca"))))'
PRRA
$ printf %s\\n AAA:K AAC:N AAG:K AAT:N ACA:T ACC:T ACG:T ACT:T AGA:R AGC:S AGG:R AGT:S ATA:I ATC:I ATG:M ATT:I CAA:Q CAC:H CAG:Q CAT:H CCA:P CCC:P CCG:P CCT:P CGA:R CGC:R CGG:R CGT:R CTA:L CTC:L CTG:L CTT:L GAA:E GAC:D GAG:E GAT:D GCA:A GCC:A GCG:A GCT:A GGA:G GGC:G GGG:G GGT:G GTA:V GTC:V GTG:V GTT:V TAA:\* TAC:Y TAG:\* TAT:Y TCA:S TCC:S TCG:S TCT:S TGA:\* TGC:C TGG:W TGT:C TTA:L TTC:F TTG:L TTT:F|tr : \ >codontable
$ echo cctcggcgggca|awk 'NR==FNR{a[tolower($1)]=$2;next}{o="";for(i=1;i<length($0);i+=3)o=o a[tolower(substr($0,i,3))];print o}' codontable -
PRRA
$ seqkit translate -l 1 # list standard table (table number 1)
The Standard Code (transl_table=1)
Source: https://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/index.cgi?chapter=tgencodes#SG1
Initiation Codons:
ATG, CTG, TTG
Stop Codons:
TAA, TAG, TGA
Stranslate Table:
AAA: K, AAC: N, AAG: K, AAT: N
ACA: T, ACC: T, ACG: T, ACT: T
AGA: R, AGC: S, AGG: R, AGT: S
ATA: I, ATC: I, ATG: M, ATT: I
CAA: Q, CAC: H, CAG: Q, CAT: H
CCA: P, CCC: P, CCG: P, CCT: P
CGA: R, CGC: R, CGG: R, CGT: R
CTA: L, CTC: L, CTG: L, CTT: L
GAA: E, GAC: D, GAG: E, GAT: D
GCA: A, GCC: A, GCG: A, GCT: A
GGA: G, GGC: G, GGG: G, GGT: G
GTA: V, GTC: V, GTG: V, GTT: V
TAA: *, TAC: Y, TAG: *, TAT: Y
TCA: S, TCC: S, TCG: S, TCT: S
TGA: *, TGC: C, TGG: W, TGT: C
TTA: L, TTC: F, TTG: L, TTT: F
snp-dists and bio3d::seqidentity only count
nucleotide changes but not insertions or deletions, so that they ignore
positions where one sequence has a gap and another sequence does not.
Biostrings::stringDist(method="hamming") also counts
positions where one sequence has a gap but another sequence does
not:
$ printf %s\\n \>a gattaca \>b aatt--->test.fa
$ snp-dists -qb test.fa
a b
a 0 1
b 1 0
$ Rscript -e 'bio3d::seqidentity(bio3d::read.fasta("test.fa"))'
a b
a 1.00 0.75
b 0.75 1.00
$ Rscript -e 'as.matrix(Biostrings::stringDist(Biostrings::readDNAStringSet("test.fa"),"hamming"))'
a b
a 0 4
b 4 0
The genomes of viruses are often missing a piece from the beginning or end of the genome, so when you have a set of aligned sequences where some sequences have gaps at either end and other sequences don't, it's common to cut out the parts from the ends of the alignment which feature gaps. For example in a paper where they produced an aligned set of genomes of SARS1, they wrote: "For network analysis, an 81-bp block at the 5'-end including gaps and a 77-bp block at the 3'-end including gaps and the poly-A tails were trimmed out of the alignment, and the final alignment contains 30,169 nucleotides." [https://www.sciencedirect.com/science/article/pii/S2590053621001075]
In the code block below where I tried aligning ten random genomes of SARS1, only three out of ten sequences included the poly(A) tail, but its length was really short in all of them: 4 bases in one sequence and 2 bases in two other sequences. The sequence with the 4-base poly-A tail was the only sequence which didn't have any gaps at the end. Two sequences had 2 gaps at the end, three sequences had 4 gaps, two sequences had 24 gaps, one sequence had 45 gaps, and one sequence had 47 gaps. At the beginning of the aligned set of sequences, six sequences had no gaps, one sequence had 20 gaps, one sequence had 32 gaps, and two sequences had 40 gaps:
$ curl -Lso sarslike.fa 'https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5'
$ seqkit seq -w0 sarslike.fa|paste - -|grep SARS\ coronavirus|gshuf --random-source=<(seq 1000)|head -n10|tr \\t \\n|mafft --quiet ->random.fa
$ mafft --clustalout random.fa # `--clustalout` uses a human-readable output format
[... lines before the beginning of the alignment omitted]
EU371559.1 atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgtt
MK062180.1 atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgtt
AY502926.1 atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgtt
FJ882926.1 --------------------------------accaacctcgatctcttgtagatctgtt
AY502931.1 atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgtt
FJ882938.1 --------------------caggaaaagccaaccaacctcgatctcttgtagatctgtt
AP006557.1 atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgtt
HQ890532.1 ----------------------------------------cgatctcttgtagatctgtt
AY321118.1 atattaggtttttacctacccaggaaaagccaaccaacctcgatctcttgtagatctgtt
HQ890535.1 ----------------------------------------cgatctcttgtagatctgtt
********************
[... lines in the middle of the alignment omitted]
EU371559.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
MK062180.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
AY502926.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
FJ882926.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
AY502931.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
FJ882938.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
AP006557.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
HQ890532.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtag---------------
AY321118.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagtgctatccccatgtg
HQ890535.1 ctgcctatatggaagagccctaatgtgtaaaattaattttagtagt--------------
*********************************************
EU371559.1 attttaatagcttcttaggagaatgacaaaa
MK062180.1 attttaatagcttcttaggagaatgac----
AY502926.1 attttaatagcttcttaggagaatgacaa--
FJ882926.1 attttaa------------------------
AY502931.1 attttaatagcttcttaggagaatgacaa--
FJ882938.1 attttaa------------------------
AP006557.1 attttaatagcttcttaggagaatgac----
HQ890532.1 -------------------------------
AY321118.1 attttaatagcttcttaggagaatgac----
HQ890535.1 -------------------------------
When I used Biostrings::stringDist(method="hamming") to
calculate a distance matrix between the ten aligned sequences, the
maximum distance was at first 120 when I didn't remove any bases, but
the maximum distance decreased to 36 when I removed the last 47 and the
first 40 bases of the alignment. When I used snp-dists to
calculate the distance matrix, the maximum distance was only 34 because
snp-dists ignores positions where one sequence has a gap
and another one doesn't:
$ Rscript -e 'max(Biostrings::stringDist(Biostrings::readDNAStringSet("random.fa"),"hamming"))'
[1] 120
$ seqkit subseq -r 41:-48 random.fa>random.trim;Rscript -e 'max(Biostrings::stringDist(Biostrings::readDNAStringSet("random.trim"),"hamming"))'
[1] 36
$ snp-dists -q random.fa|sed 1d|cut -f2-|tr \\t \\n|sort -n|tail -n1
34
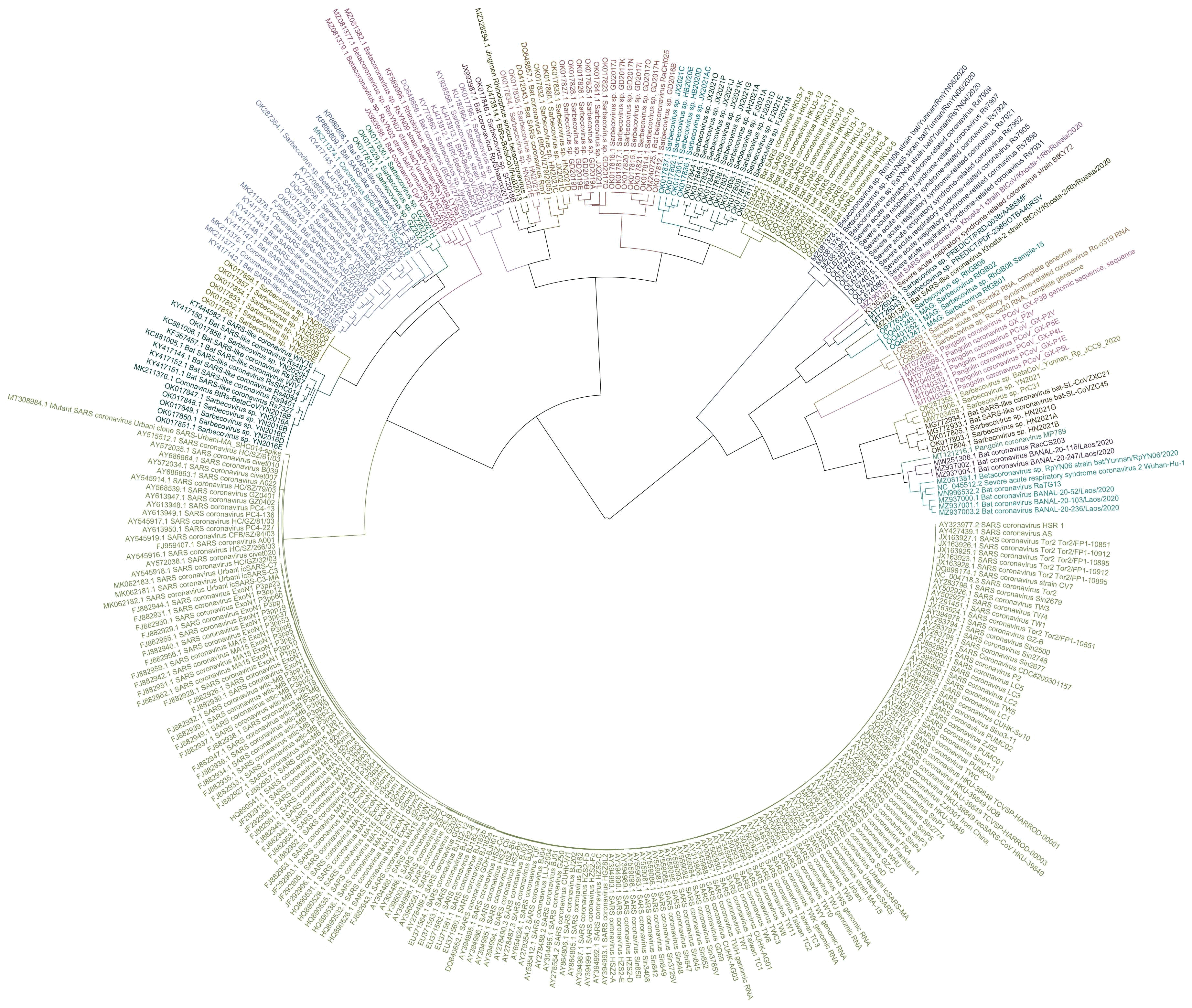
When you have a set of data that can be represented as a distance matrix, a convenient way to reorder items within the dataset so that similar items are grouped together is to order the items based on their position in a hierarchical clustering tree. I already used that method to order the sarslike.fa file I have used as an example in this thread, so in this section I'll use a file of HIV and SIV genomes as an example. The HIV Sequence Database of the Los Alamos National Laboratory has a convenient interface where you can download prealigned sets of HIV and SIV genomes. Go here: https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html. Set "Alignment type" to "Compendium" in order to download a small curated set of sequences. Set "Organism" to "Other SIV (includes HIV1 and HIV2 sequences)". Set "Region" to "GENOME" to download the whole genome. Then click "Get Alignment" and click "Download this alignment". Then run this:
library(Biostrings)
fa=readDNAStringSet("SIV_COM_2021_genome_DNA.fasta")
fa=subseq(fa,101,width(fa[1])-100)
d=stringDist(fa,"hamming")
hc=as.hclust(reorder(as.dendrogram(hclust(d)),cmdscale(as.matrix(d))[,1])) # reorder the clustering tree based on the first dimension in a multidimensional scaling matrix
writeXStringSet(fa[hc$order],"sorted.fa")
From the output, you'll see that HIV-2 samples are grouped together with SIV samples from sooty mangabey monkeys (SMM), and chimpanzee strains of SIV are clustered together with HIV-1.
The default method used by Biostrings::stringDist is
Levenshtein edit distance, which is extremely slow to calculate and
which is not needed with sequences that have already been aligned, but
the Hamming method simply returns the number of positions that are
different between two sequences.
The code above omits the first 100 and last 100 bases from distance
calculation because many samples are missing a piece from the beginning
or end of the sequence. However actually in the aligned set of HIV and
SIV genomes, many sequences are missing more than 100 bases from the
beginning or end of the alignment, so maybe it would've been better to
calculate the distance matrix between the genomes so that I only count
nucleotide changes but ignore insertions and deletions, like what's done
by bio3d::seqidentity and snp-dists.
I tried aligning 194 sequences of SARS1 and then printing the last 100 bases of the alignment:
$ curl -Lso sarslike.fa 'https://drive.google.com/uc?export=download&id=1eq9q_6jPbjvfvKnpAWDkbIyjgky7cjv5'
$ seqkit grep -nrp SARS\ coronavirus sarslike.fa|mafft --quiet --thread 4 ->sars1.aln
$ seqkit subseq -r-100:-1 sars1.aln|seqkit fx2tab|awk -F\\t '{print$2,$1}'|sed 's/, complete genome//'
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882959.1 SARS coronavirus MA15 ExoN1 isolate P3pp6
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882951.1 SARS coronavirus MA15 ExoN1 isolate P3pp3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882962.1 SARS coronavirus MA15 ExoN1 isolate P3pp10
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882942.1 SARS coronavirus MA15 ExoN1 isolate P3pp5
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882943.1 SARS coronavirus MA15 ExoN1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882957.1 SARS coronavirus MA15
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882952.1 SARS coronavirus MA15 isolate P3pp4
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882958.1 SARS coronavirus MA15 isolate P3pp7
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882948.1 SARS coronavirus MA15 isolate P3pp3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882945.1 SARS coronavirus MA15 isolate P3pp6
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- FJ882961.1 SARS coronavirus MA15 isolate P3pp5
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- MK062180.1 SARS coronavirus Urbani isolate icSARS-MA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaa-------------------------------------------------------- FJ882930.1 SARS coronavirus ExoN1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaa-------------------------------------------------------- FJ882926.1 SARS coronavirus ExoN1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- MK062179.1 SARS coronavirus Urbani isolate icSARS
tgtgtaaaattaattttagtagtgctatccccatgtgattttaa-------------------------------------------------------- FJ882938.1 SARS coronavirus wtic-MB
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- HQ890526.1 SARS coronavirus MA15 ExoN1 isolate d2ym1
tgtgtaaaattaattttagtag------------------------------------------------------------------------------ HQ890532.1 SARS coronavirus MA15 ExoN1 isolate d4ym2
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- HQ890538.1 SARS coronavirus MA15 ExoN1 isolate d2om5
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- HQ890535.1 SARS coronavirus MA15 ExoN1 isolate d2om2
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- HQ890529.1 SARS coronavirus MA15 ExoN1 isolate d2ym4
tgtgtaaaattaattttagtag------------------------------------------------------------------------------ HQ890531.1 SARS coronavirus MA15 ExoN1 isolate d4ym1
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JF292906.1 SARS coronavirus MA15 ExoN1 isolate d3om5
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JF292905.1 SARS coronavirus MA15 ExoN1 isolate d3om4
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- DQ497008.1 SARS coronavirus strain MA-15
tgtgtaaaattaattttagtag------------------------------------------------------------------------------ JF292903.1 SARS coronavirus MA15 ExoN1 isolate d4ym5
tgtgtaaaattaattttagtagtgctatccccatgtgattttaa-------------------------------------------------------- FJ882928.1 SARS coronavirus ExoN1 isolate P1pp1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaa-------------------------------------------------------- FJ882927.1 SARS coronavirus wtic-MB isolate P1pp1
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JF292909.1 SARS coronavirus MA15 isolate d2ym4
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JF292915.1 SARS coronavirus MA15 isolate d4ym5
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- HQ890541.1 SARS coronavirus MA15 isolate d2ym1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY278741.1 SARS coronavirus Urbani
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882953.1 SARS coronavirus MA15 ExoN1 isolate P3pp4
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882933.1 SARS coronavirus wtic-MB isolate P3pp6
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882935.1 SARS coronavirus wtic-MB isolate P3pp21
tgtgtaaaattaattttagtagtg---------------------------------------------------------------------------- FJ882934.1 SARS coronavirus wtic-MB isolate P3pp29
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882936.1 SARS coronavirus wtic-MB isolate P3pp2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY323977.2 SARS coronavirus HSR 1
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882937.1 SARS coronavirus wtic-MB isolate P3pp18
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- DQ898174.1 SARS coronavirus strain CV7
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY291451.1 SARS coronavirus TW1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502928.1 SARS coronavirus TW5
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882949.1 SARS coronavirus wtic-MB isolate P3pp23
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882939.1 SARS coronavirus wtic-MB isolate P3pp16
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882932.1 SARS coronavirus wtic-MB isolate P3pp14
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- JQ316196.1 SARS coronavirus HKU-39849 isolate UOB
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- NC_004718.3 SARS coronavirus Tor2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY427439.1 SARS coronavirus AS
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY283794.1 SARS coronavirus Sin2500
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- EU371559.1 SARS coronavirus ZJ02
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502929.1 SARS coronavirus TW6
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502927.1 SARS coronavirus TW4
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502926.1 SARS coronavirus TW3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394998.1 SARS coronavirus LC1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY362699.1 SARS coronavirus TWC3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502931.1 SARS coronavirus TW8
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502930.1 SARS coronavirus TW7
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY282752.2 SARS coronavirus CUHK-Su10
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AP006557.1 SARS coronavirus TWH genomic RNA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY283796.1 SARS coronavirus Sin2679
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY350750.1 SARS coronavirus PUMC01
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY321118.1 SARS coronavirus TWC
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY345986.1 SARS coronavirus CUHK-AG01
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502932.1 SARS coronavirus TW9
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaa-------------------- AY485278.1 SARS coronavirus Sino3-11
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- JN854286.1 SARS coronavirus HKU-39849 isolate recSARS-CoV HKU-39849
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502923.1 SARS coronavirus TW10
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AP006561.1 SARS coronavirus TWY genomic RNA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AP006560.1 SARS coronavirus TWS genomic RNA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY283798.2 SARS coronavirus Sin2774
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882940.1 SARS coronavirus ExoN1 isolate P3pp37
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaaaaa-------- AY357075.1 SARS coronavirus PUMC02
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY864805.1 SARS coronavirus BJ162
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY291315.1 SARS coronavirus Frankfurt 1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394990.1 SARS coronavirus HZS2-E
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394989.1 SARS coronavirus HZS2-D
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaa---------------------- AY310120.1 SARS coronavirus FRA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY345988.1 SARS coronavirus CUHK-AG03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AP006559.1 SARS coronavirus TWK genomic RNA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgaca---------------------------------- AY559081.1 SARS coronavirus Sin842
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394991.1 SARS coronavirus HZS2-Fc
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaa------------------------------ AY559087.1 SARS coronavirus Sin3725V
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AP006558.1 SARS coronavirus TWJ genomic RNA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- MK062182.1 SARS coronavirus Urbani isolate icSARS-C3-MA
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgaca---------------------------------- AY559096.1 SARS coronavirus Sin850
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394993.1 SARS coronavirus HGZ8L2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394992.1 SARS coronavirus HZS2-C
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaa-------------------- AY278491.2 SARS coronavirus HKU-39849
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY283797.1 SARS coronavirus Sin2748
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY278554.2 SARS coronavirus CUHK-W1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa AY357076.1 SARS coronavirus PUMC03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaa------------------ AY485277.1 SARS coronavirus Sino1-11
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882955.1 SARS coronavirus ExoN1 isolate P3pp19
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY864806.1 SARS coronavirus BJ202
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgaca---------------------------------- AY559088.1 SARS coronavirus SinP1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgaca---------------------------------- AY559092.1 SARS coronavirus SinP5
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882929.1 SARS coronavirus ExoN1 isolate P3pp1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaa------------------------ AY559084.1 SARS coronavirus Sin3765V
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882931.1 SARS coronavirus ExoN1 isolate P3pp12
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153547.1 Bat SARS coronavirus HKU3-12
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- DQ084199.1 bat SARS coronavirus HKU3-2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- DQ022305.2 Bat SARS coronavirus HKU3-1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- DQ084200.1 bat SARS coronavirus HKU3-3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153540.1 Bat SARS coronavirus HKU3-5
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153539.1 Bat SARS coronavirus HKU3-4
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153541.1 Bat SARS coronavirus HKU3-6
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153542.1 Bat SARS coronavirus HKU3-7
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153546.1 Bat SARS coronavirus HKU3-11
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153544.1 Bat SARS coronavirus HKU3-9
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153545.1 Bat SARS coronavirus HKU3-10
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153548.1 Bat SARS coronavirus HKU3-13
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- GQ153543.1 Bat SARS coronavirus HKU3-8
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaa------------------- DQ412043.1 Bat SARS coronavirus Rm1
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacgaaaaaaaaaaaaaaaaaaa--------------- DQ071615.1 Bat SARS coronavirus Rp3
---------------------------------------------------------------------------------------------------- AY348314.1 SARS coronavirus Taiwan TC3
---------------------------------------------------------------------------------------------------- AY338174.1 SARS coronavirus Taiwan TC1
---------------------------------------------------------------------------------------------------- AY338175.1 SARS coronavirus Taiwan TC2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY502924.1 SARS coronavirus TW11
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaa------------------- AY313906.1 SARS coronavirus GD69
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatcac----------------------------------- AY304486.1 SARS coronavirus SZ3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY395003.1 SARS coronavirus ZS-C
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394996.1 SARS coronavirus ZS-B
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- AY390556.1 SARS coronavirus GZ02
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatc------------------------------------- AY515512.1 SARS coronavirus HC/SZ/61/03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394995.1 SARS coronavirus HSZ-Cc
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394994.1 SARS coronavirus HSZ-Bc
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394987.1 SARS coronavirus HZS2-Fb
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394986.1 SARS coronavirus HSZ-Cb
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394985.1 SARS coronavirus HSZ-Bb
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaa------------------ AY278490.3 SARS coronavirus BJ03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394983.1 SARS coronavirus HSZ2-A
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaa------------------ AY278488.2 SARS coronavirus BJ01
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JX163927.1 SARS coronavirus Tor2 isolate Tor2/FP1-10851
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JX163926.1 SARS coronavirus Tor2 isolate Tor2/FP1-10912
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JX163925.1 SARS coronavirus Tor2 isolate Tor2/FP1-10895
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JX163924.1 SARS coronavirus Tor2 isolate Tor2/FP1-10851
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JX163923.1 SARS coronavirus Tor2 isolate Tor2/FP1-10912
tgtgtaaaattaattttagtagtgctatccccatgtga-------------------------------------------------------------- FJ882963.1 SARS coronavirus P2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- EU371564.1 SARS coronavirus BJ182-12
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- EU371563.1 SARS coronavirus BJ182-8
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- EU371561.1 SARS coronavirus BJ182b
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- EU371560.1 SARS coronavirus BJ182a
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY559095.1 SARS coronavirus Sin847
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatga------------------------------------ AY559086.1 SARS coronavirus Sin849
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaa--------------------------------- AY559085.1 SARS coronavirus Sin848
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaa----------------------------- AY559083.1 SARS coronavirus Sin3408
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaa---------------- AY654624.1 SARS coronavirus TJF
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- JX163928.1 SARS coronavirus Tor2 isolate Tor2/FP1-10895
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaa------------------------------ AY595412.1 SARS coronavirus LLJ-2004
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY395002.1 SARS coronavirus LC5
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY395000.1 SARS coronavirus LC3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaa------------------------------- EU371562.1 SARS coronavirus BJ182-4
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttctagg-aagaatga------------------------------------ AY559091.1 SARS coronavirus SinP4
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- MK062183.1 SARS coronavirus Urbani isolate icSARS-C7
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- MK062181.1 SARS coronavirus Urbani isolate icSARS-C3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaa----------------- AY278487.3 SARS coronavirus BJ02
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaa-------------- AY279354.2 SARS coronavirus BJ04
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttctaggagaaaatgaca---------------------------------- AY559090.1 SARS coronavirus SinP3
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttctta---------------------------------------------- AY304488.1 SARS coronavirus SZ16
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgaca---------------------------------- AY559093.1 SARS coronavirus Sin845
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgaca---------------------------------- AY559082.1 SARS coronavirus Sin852
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882947.1 SARS coronavirus wtic-MB isolate P3pp7
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882950.1 SARS coronavirus ExoN1 isolate P3pp60
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- FJ882944.1 SARS coronavirus ExoN1 isolate P3pp23
tgtgtaaaattaattttagta------------------------------------------------------------------------------- FJ882956.1 SARS coronavirus ExoN1 isolate P3pp53
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttctta---------------------------------------------- AY545914.1 SARS coronavirus isolate HC/SZ/79/03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatcacaaaaaaaaaaaaaaaa------------------- AY545918.1 SARS coronavirus isolate HC/GZ/32/03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaa------------------------------ AY545919.1 SARS coronavirus isolate CFB/SZ/94/03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatcacaaaaaaaaaaaaaaaaaaaaa-------------- AY545917.1 SARS coronavirus isolate HC/GZ/81/03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatcac----------------------------------- AY545916.1 SARS coronavirus isolate HC/SZ/266/03
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatc------------------------------------- AY572038.1 SARS coronavirus civet020
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY613948.1 SARS coronavirus PC4-13
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY613950.1 SARS coronavirus PC4-227
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY613949.1 SARS coronavirus PC4-136
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY613947.1 SARS coronavirus GZ0402
---------------------------------------------------------------------------------------------------- AY572034.1 SARS coronavirus civet007
---------------------------------------------------------------------------------------------------- AY686864.1 SARS coronavirus B039
---------------------------------------------------------------------------------------------------- AY572035.1 SARS coronavirus civet010
---------------------------------------------------------------------------------------------------- AY686863.1 SARS coronavirus A022
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaa------------------ AY278489.2 SARS coronavirus GD01
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatcacaaaaaaaa--------------------------- AY304495.1 SARS coronavirus GZ50
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gaga---------------------------------------- DQ182595.1 SARS coronavirus ZJ0301 from China
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- GU553363.1 SARS coronavirus HKU-39849 isolate TCVSP-HARROD-00001
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaa---------------------- DQ640652.1 SARS coronavirus GDH-BJH01
tgtgtaaaattaattttagtagtgctatccccatgtgatttta----cacattag-ggctcttccatatagg---------------------------- AY461660.1 SARS coronavirus SoD
tgtgtaaaattaattttagtagt----------------------------------------------------------------------------- GU553365.1 SARS coronavirus HKU-39849 isolate TCVSP-HARROD-00003
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY568539.1 SARS coronavirus GZ0401
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatcac----------------------------------- FJ959407.1 SARS coronavirus isolate A001
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394999.1 SARS coronavirus LC2
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- AY283795.1 SARS coronavirus Sin2677
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaa-------------------------------- AY394850.2 SARS coronavirus WHU
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgac----------------------------------- MT308984.1 Mutant SARS coronavirus Urbani clone SARS-Urbani-MA_SHC014-spike
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394978.1 SARS coronavirus GZ-B
tgtgtaaaattaattttagtagtgctatccccatgtgattttaatagcttcttag-gagaatgacaaaaaaaaaaaaaaaaaaaaaaaa----------- AY394979.1 SARS coronavirus GZ-C
There was only one sequence which didn't have any gaps at the end, but the number of gaps at the end ranged from 8 to 242 in the other sequences, where in over half of the cases the only difference was the length of the poly(A) tail.
From the tenth-last line of the output above, you can see that AY461660 (SARS coronavirus SoD) has a 24-base segment at the end which is not shared by any other sequence in my alignment (CACATTAGGGCTCTTCCATATAGG). However the 24-base segment turned out to be the reverse complement of another 24-base segment near the end of the genome which started 63 bases earlier:
$ seqkit seq -g sars1.aln|seqkit grep -nrp AY461660|seqkit locate -p cacattagggctcttccatatagg|awk '{$2=""}1'|column -t
seqID pattern strand start end matched
AY461660.1 cacattagggctcttccatatagg + 29692 29715 cacattagggctcttccatatagg
AY461660.1 cacattagggctcttccatatagg - 29629 29652 cacattagggctcttccatatagg
$ seqkit seq -g sars1.aln|seqkit grep -nrp AY461660|seqkit subseq -r29629:29652 # print the region of the reverse complement
>AY461660.1 SARS coronavirus SoD, complete genome
cctatatggaagagccctaatgtg
seqkit locate doesn't ignore gaps so I used
seqkit seq -g to delete gaps. Another way to delete gaps is
sed '/^>/!s/-//g'.
When I also looked at the first 100 bases of each sequence, I noticed another assembly error which was that in the beginning of AY559083 (SARS coronavirus Sin3408), there's two copies of a 29-base segment (GGAAAAGCCAACCAACCTCGATCTCTTGT), where one copy starts at position 22 and another copy starts at position 57:
$ seqkit seq -g sars1.aln|seqkit grep -nrp AY559083|seqkit locate -p ggaaaagccaaccaacctcgatctcttgt|awk '{$2=""}1'|column -t
seqID pattern strand start end matched
AY559083.1 ggaaaagccaaccaacctcgatctcttgt + 22 50 ggaaaagccaaccaacctcgatctcttgt
AY559083.1 ggaaaagccaaccaacctcgatctcttgt + 57 85 ggaaaagccaaccaacctcgatctcttgt
See https://docs.nextstrain.org/projects/nextclade/en/stable/user/nextclade-cli.html.
$ brew install --cask miniconda;conda init ${SHELL##*/} # this didn't work without `--cask`
[...]
$ conda install -c bioconda nextclade
[...]
$ nextclade dataset get --name sars-cov-2 --output-dir sars-cov-2 # download files required to analyze SARS2 variants
[...]
$ nextclade run --input-dataset sars-cov-2 --output-all outdir sars-cov-2/sequences.fasta # `sequences.fasta` is a demo dataset of SARS2 sequences
[...]
$ awk -F\\t 'NR>1{print$2": "$8" ("$17" substitutions, "$18" insertions, "$19" deletions)"}' outdir/nextclade.tsv|tail -n3
USA/MA-CDCBI-CRSP_6WUXOVDJ3B5G2RNQ/2021: 21J (Delta) (32 substitutions, 4 insertions, 0 deletions)
USA/CA-CDC-FG-161774/2021: 21J (Delta) (34 substitutions, 4 insertions, 0 deletions)
OU819939: 21J (Delta) (31 substitutions, 6 insertions, 0 deletions)
$ head -n2 outdir/nextclade.tsv|csvtk -t transpose|column -s$'\t' -t # show the results of one sample in the demo dataset
index 1
seqName OP971202
clade 23A
Nextclade_pango XBB.1.5.13
partiallyAliased XBB.1.5.13
clade_nextstrain 23A
clade_who Omicron
clade_legacy 23A (Omicron)
qc.overallScore 0
qc.overallStatus good
totalSubstitutions 92
totalDeletions 30
totalInsertions 0
totalFrameShifts 0
totalMissing 0
totalNonACGTNs 0
totalAminoacidSubstitutions 65
totalAminoacidDeletions 13
totalAminoacidInsertions 0
totalUnknownAa 0
alignmentScore 88550
alignmentStart 55
alignmentEnd 29733
coverage 0.992509112798047
isReverseComplement false
substitutions C241T,A405G,T670G,C2790T,C3037T,G4184A,C4321T,C9344T,A9424G,C9534T,C9866T,C10029T,C10198T,G10447A,C10449A,C12880T,C14408T,G15451A,C15714T,C15738T,T15939C,T16342C,T17124C,C17410T,T17859C,A18163G,A19326G,C19955T,A20055G,C21618T,T21810C,T21880C,G21987A,C22000A,C22109G,T22200A,G22317T,G22577C,G22578A,G22599C,C22664A,C22674T,T22679C,C22686T,A22688G,G22775A,A22786C,G22813T,T22882G,G22895C,T22896C,G22898A,T22942G,G22992A,C22995A,A23013C,T23018C,T23019C,T23031C,A23055G,A23063T,T23075C,A23403G,C23525T,T23599G,C23604A,C23854A,G23948T,A24424T,T24469A,C25000T,C25416T,C25584T,C26060T,C26270T,A26275G,C26577G,G26709A,C26858T,A27259C,G27382C,A27383T,T27384C,C27807T,G27915T,G28079T,A28271T,C28311T,G28881A,G28882A,G28883C,A29510C
deletions 11288-11296,21633-21641,21992-21994,28362-28370
insertions
frameShifts
aaSubstitutions E:T9I,E:T11A,M:Q19E,M:A63T,N:P13L,N:R203K,N:G204R,N:S413R,ORF1a:K47R,ORF1a:S135R,ORF1a:T842I,ORF1a:G1307S,ORF1a:L3027F,ORF1a:T3090I,ORF1a:L3201F,ORF1a:T3255I,ORF1a:P3395H,ORF1b:P314L,ORF1b:G662S,ORF1b:S959P,ORF1b:R1315C,ORF1b:I1566V,ORF1b:T2163I,ORF3a:T223I,ORF6:D61L,ORF8:G8*,ORF9b:P10S,S:T19I,S:A27S,S:V83A,S:G142D,S:H146Q,S:Q183E,S:V213E,S:G252V,S:G339H,S:R346T,S:L368I,S:S371F,S:S373P,S:S375F,S:T376A,S:D405N,S:R408S,S:K417N,S:N440K,S:V445P,S:G446S,S:N460K,S:S477N,S:T478K,S:E484A,S:F486P,S:F490S,S:Q498R,S:N501Y,S:Y505H,S:D614G,S:H655Y,S:N679K,S:P681H,S:N764K,S:D796Y,S:Q954H,S:N969K
aaDeletions N:E31-,N:R32-,N:S33-,ORF1a:S3675-,ORF1a:G3676-,ORF1a:F3677-,ORF9b:E27-,ORF9b:N28-,ORF9b:A29-,S:L24-,S:P25-,S:P26-,S:Y144-
aaInsertions
privateNucMutations.reversionSubstitutions
privateNucMutations.labeledSubstitutions
privateNucMutations.unlabeledSubstitutions
privateNucMutations.totalReversionSubstitutions 0
privateNucMutations.totalLabeledSubstitutions 0
privateNucMutations.totalUnlabeledSubstitutions 0
privateNucMutations.totalPrivateSubstitutions 0
missing
unknownAaRanges
nonACGTNs
qc.missingData.missingDataThreshold 3000
qc.missingData.score 0
qc.missingData.status good
qc.missingData.totalMissing 0
qc.mixedSites.mixedSitesThreshold 10
qc.mixedSites.score 0
qc.mixedSites.status good
qc.mixedSites.totalMixedSites 0
qc.privateMutations.cutoff 24
qc.privateMutations.excess -8
qc.privateMutations.score 0
qc.privateMutations.status good
qc.privateMutations.total 0
qc.snpClusters.clusteredSNPs
qc.snpClusters.score 0
qc.snpClusters.status good
qc.snpClusters.totalSNPs 0
qc.frameShifts.frameShifts
qc.frameShifts.totalFrameShifts 0
qc.frameShifts.frameShiftsIgnored
qc.frameShifts.totalFrameShiftsIgnored 0
qc.frameShifts.score 0
qc.frameShifts.status good
qc.stopCodons.stopCodons
qc.stopCodons.totalStopCodons 0
qc.stopCodons.score 0
qc.stopCodons.status good
totalPcrPrimerChanges 7
pcrPrimerChanges Charité_RdRp_F:G15451A,Charité_E_F:C26270T;A26275G,ChinaCDC_N_F:G28881A;G28882A;G28883C,USCDC_N1_P:C28311T
failedGenes
warnings
errors
Nextclade also produces a FASTA file where each sequence is aligned against Wuhan-Hu-1, and it additionally produces an aligned FASTA file for each protein. Below you can see that among the samples included in the demo dataset, the QTNSPRRA motif of the spike protein has changed to QTNSRRRA in most delta samples and to QTKSHRRA in most Omicron samples:
$ seqkit subseq -r $(seqkit locate -p QTNSPRRA outdir/nextclade_gene_S.translation.fasta|awk 'NR==2{print$5":"$6}') outdir/nextclade_gene_S.translation.fasta|grep -v \>|sort|uniq -c|sort -r
37 QTNSPRRA
32 QTKSHRRA
27 QTNSRRRA
18 QTNSHRRA
2 XXXXXXXX
1 QTNXXXXX
1 QTNSPRXX
1 QTNSPRRX
1 QTKSHRRX
1 HTNSPRXX
1 HTNSPRRA
$ seqkit subseq -r $(seqkit locate -p QTNSPRRA outdir/nextclade_gene_S.translation.fasta|awk 'NR==2{print$5":"$6}') outdir/nextclade_gene_S.translation.fasta|seqkit grep -vsp X|seqkit fx2tab|awk -F\\t 'NR==FNR{a[$1]=$2;next}{print$2" "a[$1]}' <(awk -F\\t 'NR>1{print$2"\t"$8}' outdir/nextclade.tsv) -|sort|uniq -c|sort -r
18 QTNSRRRA 21J (Delta)
7 QTNSHRRA 20I (Alpha, V1)
5 QTKSHRRA 23A (Omicron)
5 QTKSHRRA 21L (Omicron)
4 QTNSPRRA 20A
4 QTKSHRRA recombinant
4 QTKSHRRA 22D (Omicron)
4 QTKSHRRA 22B (Omicron)
3 QTNSRRRA 21I (Delta)
3 QTNSRRRA 21B (Kappa)
3 QTNSRRRA 21A (Delta)
3 QTNSPRRA 21G (Lambda)
3 QTNSPRRA 21F (Iota)
3 QTNSPRRA 20J (Gamma, V3)
3 QTNSPRRA 20H (Beta, V2)
3 QTNSPRRA 20G
3 QTNSPRRA 20F
3 QTNSPRRA 20E (EU1)
3 QTNSPRRA 20D
3 QTNSPRRA 19B
3 QTNSHRRA 21H (Mu)
3 QTNSHRRA 21E (Theta)
3 QTNSHRRA 20B
3 QTKSHRRA 21K (Omicron)
2 QTNSPRRA 21C (Epsilon)
2 QTNSPRRA 20C
2 QTNSHRRA recombinant
2 QTKSHRRA 22C (Omicron)
2 QTKSHRRA 22A (Omicron)
1 QTNSPRRA 21D (Eta)
1 QTNSPRRA 19A
1 QTKSHRRA 22F (Omicron)
1 QTKSHRRA 22E (Omicron)
1 QTKSHRRA 21M (Omicron)
1 HTNSPRRA 21D (Eta)
The SARS2 data from GISAID is not publicly available from Nextstrain but the data from GenBank is. Nextstrain's website has links to FASTA files of global and regional subsamples from GenBank. [https://docs.nextstrain.org/projects/ncov/en/latest/reference/remote_inputs.html] Each file consists of about 3000-4000 sequences.
$ curl -s https://data.nextstrain.org/files/ncov/open/global/aligned.fasta.xz|gzip -dc>global.fa # macOS gzip supports decompressing XZ
$ curl -s https://data.nextstrain.org/files/ncov/open/global/metadata.tsv.xz|gzip -dc>global.tsv
$ seqkit stat global.fa # all sequences have the same length because they are aligned against Wuhan-Hu-1
file format type num_seqs sum_len min_len avg_len max_len
global.fa FASTA DNA 2,929 87,585,887 29,903 29,903 29,903
$ awk -F\\t 'NR>1{x=substr($7,1,4);a[x]+=$37;n[x]++}END{for(i in a)print i,n[i],a[i]/n[i]}' OFMT=%.1f global.tsv|sort # show the number of sequences from each year and the average number of nucleotide changes for each year
2019 1 0
2020 280 14.1
2021 346 34.8
2022 1791 75.5
2023 512 86.9
$ sed 1d global.tsv|cut -f8|sort|uniq -c|sort # number of samples per continent
43 Oceania
217 South America
525 North America
711 Africa
715 Europe
719 Asia
$ sed 1d global.tsv|cut -f20|sort|uniq -c|sort # number of samples per variant
1
1 21E (Theta)
1 21M (Omicron)
3 20F
3 21F (Iota)
5 21C (Epsilon)
6 21H (Mu)
7 20E (EU1)
7 21A (Delta)
7 21D (Eta)
9 21G (Lambda)
13 22C (Omicron)
13 recombinant
14 20G
17 20D
19 20H (Beta, V2)
20 19B
22 20J (Gamma, V3)
23 21I (Delta)
27 19A
40 20C
50 22A (Omicron)
68 20I (Alpha, V1)
69 21L (Omicron)
80 21K (Omicron)
93 20A
103 21J (Delta)
113 20B
180 23A (Omicron)
221 22D (Omicron)
291 22F (Omicron)
569 22E (Omicron)
835 22B (Omicron)
Here's all metadata for one sample:
$ tp(){ awk '{for(i=1;i<=NF;i++)a[i][NR]=$i}END{for(i in a)for(j in a[i])printf"%s"(j==NR?"\n":FS),a[i][j]}' "FS=${1-$'\t'}";} # transpose
$ tab(){ awk '{if(NF>m)m=NF;for(i=1;i<=NF;i++){a[NR][i]=$i;l=length($i);if(l>b[i])b[i]=l}}END{for(h in a){for(i=1;i<=m;i++)printf("%-"(b[i]+n)"s",a[h][i]);print""}}' "${1+FS=$1}" "n=${2-1}"|sed 's/ *$//';} # this is like `column -s` but it tolerates empty cells and prints one instead of two spaces between columns
$ sed 2q global.tsv|tp|tab \\t
strain DJI/NAMRU3_IT001/2021
virus ncov
gisaid_epi_isl EPI_ISL_6693791
genbank_accession OL640827
genbank_accession_rev OL640827.1
sra_accession
date 2021-10-12
region Africa
country Djibouti
division Djibouti
location
region_exposure Africa
country_exposure Djibouti
division_exposure Djibouti
segment genome
length 29764
host Homo sapiens
age ?
sex ?
Nextstrain_clade 21J (Delta)
pango_lineage AY.127.1
GISAID_clade ?
originating_lab ?
submitting_lab Naval Medical Research Center, Biological Defense Research Directorate, Genomics and Bioinformatics Department
authors Arnold et al
url https://www.ncbi.nlm.nih.gov/nuccore/OL640827
title
paper_url ?
date_submitted 2021-11-26
date_updated 2021-11-26
sampling_strategy ?
database genbank
Nextclade_pango AY.127.1
immune_escape ?
ace2_binding ?
missing_data 0
divergence 43
nonACGTN 0
coverage 0.9957863759489014
rare_mutations 2
reversion_mutations 0
potential_contaminants 0
QC_missing_data good
QC_mixed_sites good
QC_rare_mutations good
QC_snp_clusters good
QC_frame_shifts good
QC_stop_codons good
QC_overall_score 0
QC_overall_status good
frame_shifts
deletions 22029-22034,28248-28253,28271
insertions
substitutions G210T,C241T,G242A,C3037T,G4181T,T4634C,C6402T,T7049C,C7124T,T7816C,C8986T,G9053T,C10029T,A11201G,A11332G,C14408T,G15451A,C16466T,C19220T,A19445G,C21618G,C21846T,G21987A,T22917G,C22995A,A23403G,C23604G,G23608T,C24138A,G24410A,C25469T,G25538T,C26054A,T26767C,T27638C,C27752T,C27874T,G28378T,A28461G,G28881T,G28916T,G29402T,G29742T
aaSubstitutions M:I82T,N:D63G,N:R203M,N:G215C,N:D377Y,ORF1a:A1306S,ORF1a:P2046L,ORF1a:Y2262H,ORF1a:P2287S,ORF1a:V2930L,ORF1a:T3255I,ORF1a:T3646A,ORF1b:P314L,ORF1b:G662S,ORF1b:P1000L,ORF1b:A1918V,ORF1b:K1993R,ORF3a:S26L,ORF3a:G49V,ORF3a:T221K,ORF7a:V82A,ORF7a:T120I,ORF7b:T40I,ORF9b:R32L,ORF9b:T60A,S:T19R,S:T95I,S:G142D,S:R158G,S:L452R,S:T478K,S:D614G,S:P681R,S:T859N,S:D950N
clock_deviation 1.00
I tried searching for samples dated 2023 with the lowest number of nucleotide changes from Wuhan-Hu-1. The first two samples below have only 3 nucleotide changes, but it's probably because they have an incorrect collection date at GenBank, because at GenBank the collection date of OQ244525 is also listed as January 3rd 2023:
$ awt '$7~/^2023/{print$37,$1,$4,$20,$7}' global.tsv|sort -n|head|tab \\t
3 OQ244525 OQ244525 19A 2023-01-03
3 OQ519925 OQ519925 19A 2023-02-02
47 OQ346308 OQ346308 22B (Omicron) 2023-01-11
51 OQ439757 OQ439757 22F (Omicron) 2023-01-27
52 OQ346309 OQ346309 recombinant 2023-01-11
57 THA/CONI-3970/2023 OQ585995 22F (Omicron) 2023-02-02
58 USA/OKPHL0025701/2023 OQ458506 23A (Omicron) 2023-01-23
61 KEN/SS11785/2023 OQ608726 22E (Omicron) 2023-01-19
62 JPN/SZ-NTC-166/2023 LC758541 22B (Omicron) 2023-02-19
62 OX416655 OX416655 22E (Omicron) 2023-01-10
There's three samples that are missing more than two thirds of all SNPs. It causes them to have only 12 nucleotide changes from Wuhan-Hu-1 even though they are Omicron samples:
$ copt(){ awk -F\\t 'NR==FNR{a[NR]=$0;next}{for(i in a)printf"%s"(i==length(a)?"\n":FS),$(a[i])}' <(printf %s\\n "$@") -; }
$ copt 36 1 20 37 <global.tsv|sort -rn|head|tab \\t
22930 Switzerland/ZH-UZH-IMV-3ba67d27/2022 21L (Omicron) 12
22930 Switzerland/ZH-UZH-IMV-3ba678a9/2022 21L (Omicron) 12
22930 Switzerland/ZH-UZH-IMV-3ba66eac/2022 21L (Omicron) 12
11898 OQ439756 22F (Omicron) 76
11437 CHN/CPL-12-43/2022 22B (Omicron) 29
11275 BGD/Laila_489/2021 21J (Delta) 25
9420 Switzerland/GR-ETHZ-150039/2020 20A 5
9109 OQ244528 22B (Omicron) 46
8945 PRY/WOR12903A608_202103275_2-4710519/2020 19B 7
8787 OQ692394 22F (Omicron) 64
This downloads NextStrain's subsamples for each continent, which you can concatenate to get a bigger global dataset:
u=https://data.nextstrain.org/files/ncov/open;for x in africa asia europe north-america oceania south-america;do curl $u/$x/aligned.fasta.xz>continent.$x.fa.xz;curl $u/$x/metadata.tsv.xz>continent.$x.tsv.xz;done
curl https://data.nextstrain.org/files/ncov/open/global/aligned.fasta.xz|gzip -dc>global.fa curl https://data.nextstrain.org/files/ncov/open/global/metadata.tsv.xz|gzip -dc>global.tsv brew install brewsci/bio/snp-dists snp-dists global.fa>global.dist
# `geom_mark_hull` draws the hulls incorrectly in new versions of `ggforce` so you need to install an old version of `ggforce`:
# `install.packages("https://cran.r-project.org/src/contrib/Archive/ggforce/ggforce_0.3.4.tar.gz")`
library(ggplot2)
library(ggrepel)
t=read.table("global.dist",header=T,row.names=1,sep="\t")
meta=read.table("global.tsv",header=T,row.names=1,sep="\t",quote="")
meta=meta[rownames(t),]
pick=meta[,35]<1000 # reject samples with 1000 or more missing SNPs
t=t[pick,pick]
meta=meta[pick,]
variant=meta$Nextstrain_clade
mds0=cmdscale(t,ncol(t)-1,eig=T) # `cmdscale(t,2,eig=T)` would be a lot faster but it doesn't return all eigenvalues so the proportion of explained variance cannot be calculated correctly
mds=as.data.frame(mds0$points)
eig=head(mds0$eig,ncol(mds))
pct=paste0("Dim ",1:ncol(mds)," (",sprintf("%.1f",100*eig/sum(eig)),"%)")
centers=data.frame(aggregate(mds,list(variant),mean),row.names=1)
k=as.factor(variant)
set.seed(0)
color=as.factor(sample(length(unique(k))))
hue=c(0,30,60,100,170,210,240,300)
cl=list(c(70,60),c(70,40),c(30,70),c(30,30))
pal1=unlist(lapply(cl,\(x)hcl(hue+15,x[1],x[2])))
pal2=unlist(lapply(cl,\(x)hcl(hue+15,if(x[2]>=60).5*x[1]else .4*x[1],if(x[2]>=60).1*x[2]else 90)))
i=1 # change to `i=3` to plot dimensions 3 and 4
xdim=as.name(paste0("V",i))
ydim=as.name(paste0("V",i+1))
# draw a line between each point and its two closest neighbors
seg=cbind(mds[rep(1:nrow(mds),each=2),c(i,i+1)],mds[apply(t,1,\(x)order(x)[2:3]),c(i,i+1)])
colnames(seg)=paste0("V",1:4)
ggplot(mds,aes(!!xdim,!!ydim),group=0)+
ggforce::geom_mark_hull(aes(group=k),color=pal1[color[k]],fill=pal1[color[k]],concavity=1000,radius=unit(.15,"cm"),expand=unit(.15,"cm"),alpha=.2,size=.15)+
geom_segment(data=seg,aes(x=V1,y=V2,xend=V3,yend=V4),color="gray75",linewidth=.15)+
geom_point(color=pal1[color[k]],size=.5)+
labs(x=pct[i],y=pct[i+1])+
# geom_label(data=centers,aes(V1,V2,label=rownames(centers)),fill=pal1[color],color=pal2[color],alpha=.8,size=2.2,label.r=unit(.1,"lines"),label.padding=unit(.1,"lines"),label.size=.1)+ # allow overlapping labels
ggrepel::geom_label_repel(data=centers,aes(!!xdim,!!ydim),fill=pal1[color],color=pal2[color],label=rownames(centers),size=2.2,alpha=.8,label.r=unit(.1,"lines"),label.padding=unit(.1,"lines"),label.size=.1,box.padding=0.1,max.overlaps=Inf,point.size=0,segment.size=.2,force=3)+
scale_x_continuous(breaks=seq(-1000,1000,20),expand=expansion(mult=c(.04,.04)))+
scale_y_continuous(breaks=seq(-1000,1000,20),expand=expansion(mult=c(.04,.04)))+
theme(
axis.ticks=element_line(linewidth=.4,color="gray80"),
axis.ticks.length=unit(-.15,"cm"),
axis.text=element_text(color="black",size=6),
axis.text.x=element_text(margin=margin(.3,0,0,0,"lines")),
axis.text.y=element_text(angle=90,vjust=1,hjust=.5,margin=margin(0,.4,0,0,"lines")),
axis.title=element_text(size=8),
legend.position="none",
panel.background=element_rect(fill="white"),
panel.border=element_rect(color="gray80",fill=NA,linewidth=.8),
panel.grid=element_blank(),
plot.background=element_rect(fill="white")
)
ggsave(paste0(i,".png"),width=7,height=7)
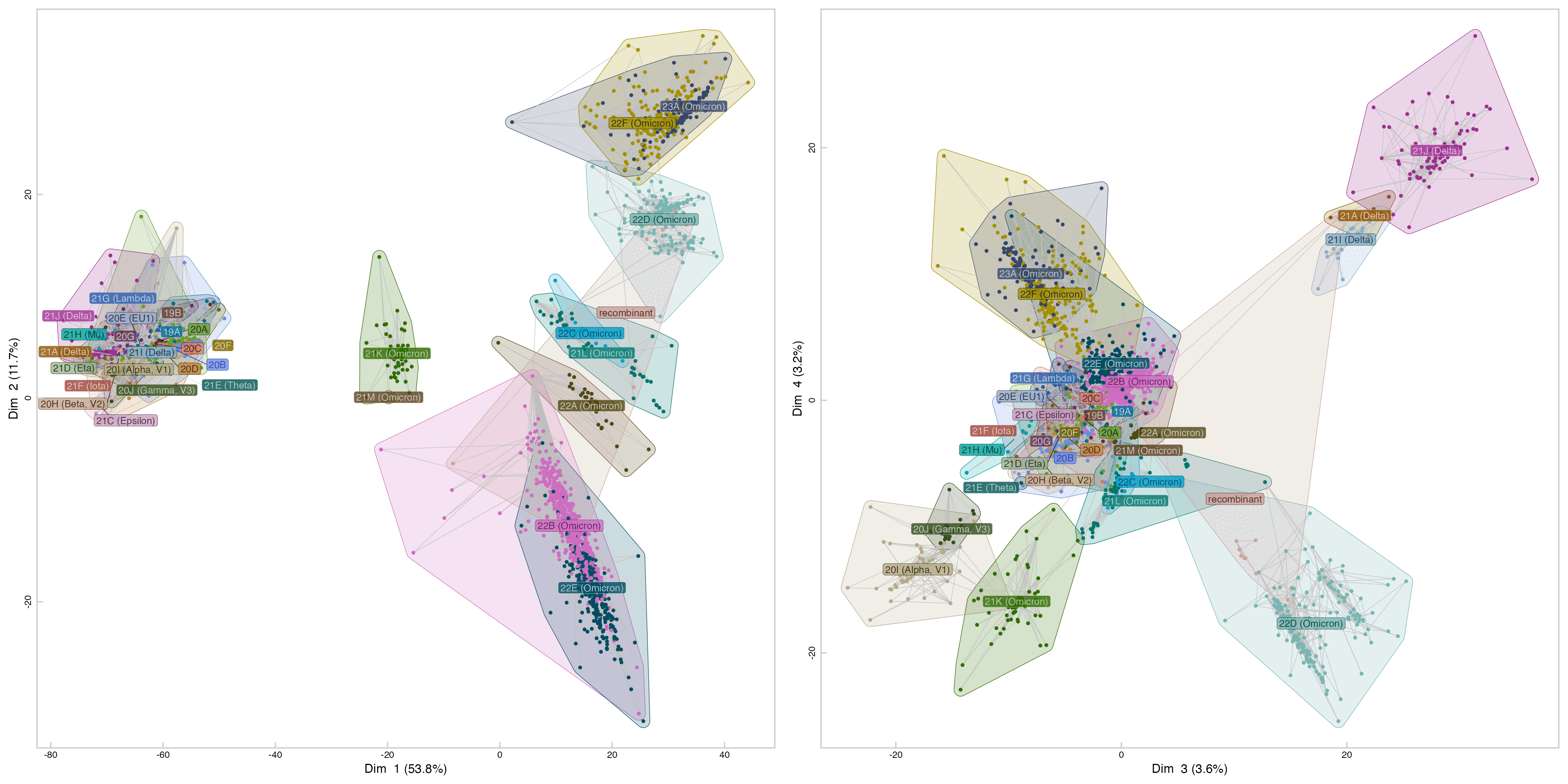
I entered the accession number of Wuhan-Hu-1's spike protein to protein BLAST (QHD43416). [https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins] I entered "SARS-CoV-2 (taxid:2697049)" in the organism field and clicked the "exclude" checkbox next to it. Then I clicked "Algorithm parameters" and I set "Max target sequences" to 1000. Then I clicked I clicked "BLAST" and I selected "FASTA (complete sequence)" from the "Download" menu.
The following code adds Wuhan-Hu-1's spike protein to the file I downloaded from BLAST, aligns the sequences, excludes sequences without a parent organism in the nuccore database, creates a TSV file for metadata about the sequences, and creates a new FASTA file with renamed sequences:
# download the SARS2 spike and align the spike proteins (`--reorder` groups similar proteins together instead of preserving the previous order)
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&rettype=fasta&id=YP_009724390'>sars2.spike.fa
cat seqdump.txt sars2.spike.fa|mafft --reorder ->spike1k.aln
# `emu` stands for "Entrez multi"
# this uses `curl -d` to send the data via a POST request, because if you specify more than a few hundred accessions at a time as part of the URL, you get an error that the URL is too long
# `xml fo -D` deletes the DOCTYPE so xmlstarlet won't later try to fetch external entities
emu(){ curl -d id=$(paste -sd, -) "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1-nuccore}&retmode=${2-xml}${3+&rettype=$3}";}
sed -n 's/^>\([^ ]*\).*/\1/p' spike1k.aln|emu protein|xml fo -D>spike1k.xml
# the full organism title is not included in the XML metadata of the protein sequences, so you need to get the organism title from the parent nucleotide sequence
# 84 out of 1001 of the spike sequences were missing the parent accession number, but they consisted of 83 entries like "pdb: molecule 7QUR, chain A" and one contaminated sequence which had been deleted from nuccore
# my code also omitted 8 entries from the UniProt database whose `GBSeq_source-db` field started with text like "UniProtKB: locus SPIKE_BC133, accession Q0Q4F2"
xml sel -t -v //GBSeq_source-db spike1k.xml|sed -En 's/^(REFSEQ: )?accession //p'|emu|xml fo -D>spike1k.nuc.xml
# get TSV metadata from protein XML
<spike1k.xml xml sel -t -m //GBSeq -v GBSeq_accession-version -o $'\t' -v .//GBSeq_source-db -o $'\t' -v './/GBQualifier[GBQualifier_name="isolate"]/GBQualifier_value' -o $'\t' -v GBSeq_definition -o $'\t' -v GBSeq_length -o $'\t' -v GBSeq_create-date -o $'\t' -v './/GBQualifier[GBQualifier_name="collection_date"]/GBQualifier_value' -o $'\t' -v './/GBQualifier[GBQualifier_name="country"]/GBQualifier_value' -o $'\t' -v './/GBReference[last()]//GBAuthor[1]' -o ... -v './/GBReference[last()]//GBAuthor[last()]' -o $'\t' -v '(.//GBReference_title[text()!="Direct Submission"])[last()]' -n>temp.tsv
# get the title of the parent organism from the nucleotide XML and make a short version of the organism title
xml sel -t -v //GBSeq_definition spike1k.nuc.xml|sed -E 's/, (complete|partial|nearly complete) (gene?ome|cds|sequence)| whole genome shotgun sequence| (spike glycoprotein|spike glycoprotein precursor|s protein|surface glycoprotein).*| (spike|s)( protein)?( \(S\))? gene| spike protein gene| S protein \(S\) gene| [A-Za-z0-9] gene;.*|gene;.*| putative .*|[oO][rR][fF]1ab?[ ,].*|; and .*| S protein gene.*| spike protein .*| S gene .*| S[12] protein.*| polyprotein .*|( genomic)? RNA$| genomic RNA.*//g;s/ isolate / /;s/Middle East respiratory syndrome/MERS/;s/Severe acute respiratory syndrome/SARS/;s/ *$//'|paste <(xml sel -t -m //GBSeq -v GBSeq_accession-version -o $'\t' -v GBSeq_definition -n spike1k.nuc.xml) ->temp2.tsv
# generate the final TSV and FASTA files
redate(){ tr A-Z a-z|sed -f <(paste -d/ <(printf s/%s\\n jan feb mar apr may jun jul aug sep oct nov dec) <(printf %s\\n {01..12}/g))|sed -E 's/(..)-(..)-(....)/\3-\2-\1/g;s/(..)-([0-9]{4})/\2-\1/g';}
paste <(cut -f-5 temp.tsv|awk '{sub("^(REFSEQ: )?accession ","",$2)}1' {,O}FS=\\t) <(cut -f6-7 temp.tsv|redate) <(cut -f8- temp.tsv)|awk -F\\t 'NR==FNR{a[$1]=$2"\t"$3;next}$2 in a{print$0"\t"a[$2]}' temp2.tsv -|cat <(printf %s\\n protein_accession organism_accession protein_title isolate_name protein_length genbank_added_date collection_date collection_country oldest_authors oldest_reference organism_title organism_short_title|paste -s -) ->spike1k.tsv
awk -F\\t '{print$1"\t"$12"|"$4"|"$6"|"$7"|"$8"|"$2"|"$10}' spike1k.tsv|awk -F\\t 'NR==FNR{a[$1]=$0;next}$1 in a{print a[$1]"\t"$2}' - <(sed 's/ .*//' spike1k.aln|seqkit fx2tab)|sed $'s/\t/ /'|seqkit tab2fx>spike1k.fa
I uploaded the FASTA file generated by the code above here: https://drive.google.com/uc?export=download&id=1riMniCLMe4Aqg-hj4O6LXQaqvx2gPnHx. And I uploaded the TSV file here: https://drive.google.com/uc?export=download&id=1o79qDLcrbxAppnK15t7tjdn7vKU2H3dT.
curl -Lso spike1k.fa 'https://drive.google.com/uc?export=download&id=1riMniCLMe4Aqg-hj4O6LXQaqvx2gPnHx'
curl -Lso spike1k.tsv 'https://drive.google.com/uc?export=download&id=1o79qDLcrbxAppnK15t7tjdn7vKU2H3dT'
# show the number of amino acid changes of the sequence whose ID is specified as an argument to each sequence from STDIN; ignore positions where either sequence has an X letter
dif2x(){ x=$(cat);awk 'NR==1{split($0,a,"");l=length;next}{split($0,b,"");n=0;for(i=1;i<=l;i++)if(a[i]!="X"&&b[i]!="X"&&a[i]!=b[i])n++;print n}' <(seqkit grep -p "$1"<<<"$x"|seqkit seq -s;seqkit seq -s<<<"$x");}
# colorize amino acids
cola(){ sed -f <(awk -F: '{print"s/"$1"/\033[38;2;0;0;0m\033[48;2;"$2";"$3";"$4"m"$1"\033[0m/g"}' <(printf %s\\n A:242:121:121 C:242:182:121 D:242:242:121 E:182:242:121 F:121:242:151 G:121:242:222 H:121:182:242 I:121:121:242 K:182:121:242 L:242:121:242 M:255:191:191 N:255:223:191 P:255:255:191 Q:223:255:191 R:191:255:207 S:191:255:244 T:191:223:255 V:191:191:255 W:223:191:255 Y:255:191:255 -:140:140:140)) "$@";}
# `tab \\t` is like `column -ts$'\t'` but it doesn't get thrown off by empty fields and you can change the number of spaces that get printed between columns
tab(){ awk '{if(NF>m)m=NF;for(i=1;i<=NF;i++){a[NR][i]=$i;l=length($i);if(l>b[i])b[i]=l}}END{for(h in a){for(i=1;i<=m;i++)printf(i==m?"%s\n":"%-"(b[i]+n)"s",a[h][i])}}' "${1+FS=$1}" "n=${2-1}";}
f=spike1k.fa;beg=1830;end=1860;targ=YP_009825051.1;sub=$(seqkit subseq -r $beg:$end $f);paste <(dif2x $targ<<<"$sub") <(dif2x $targ<$f) <(seqkit seq -n $f)|tab \\t|cut -c-$[COLUMNS-(end-beg+2)]|paste -d\ <(seqkit seq -s<<<"$sub"|cola) -
From the output, you can see that the RRA part of the PRRA insert is included in sequences like "Murine coronavirus strain JHM-WU-Dns2", "Murine coronavirus MHV-3 UNICAMP", "Pipistrellus bat coronavirus HKU5 16S", "BtPa-BetaCoV/GD2013", "Bat coronavirus HKU5-2", "Bat coronavirus HKU5-3", and "Bat coronavirus (BtCoV/A434/2005)". The second and third columns show the number of amino acid changes from Wuhan-Hu-1, where the second column includes only the displayed region and the third column includes the whole genome:
The code below downloads the first 100,000 reads from one of the two paired read files from a paper from 2020 titled "In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges". It then makes a local BLAST database of the reads and takes the 100 reads which have the best match to the first read of the file, and it aligns the 100 reads and prints the most common nucleotide value at each position of the alignment:
$ brew install blast seqkit mafft emboss
$ curl ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR445/004/ERR4451134/ERR4451134_1.fastq.gz|seqkit head -n100000|seqkit fq2fa>temp.fa
$ makeblastdb -in temp.fa -dbtype nucl
$ seqkit head -n1 temp.fa|blastn -db temp.fa -outfmt '6 qseqid sseqid pident length bitscore qlen slen stitle qstart qend sstart send'|cut -f2|seqkit grep -f- temp.fa|mafft --quiet ->temp.aln
$ seqkit fx2tab temp.aln|head -n4 # show first 4 aligned reads
ERR4451134.1 M00758:969:CG8RD:1:1102:16602:1915/1 ------------------------ctcataataattagtaatatctctgctatagtaacctgaaagtcaacgagatgaaacatctgttttctcttactgtacaagcaaagcaatattgtcactgctactggtatggtctgtgtttaatttatagttgccaatcctgtagcgact------------------------
ERR4451134.353 M00758:969:CG8RD:1:1102:18404:2089/1 -----------------------cctcataataattagtaatatctctgctatagtaacctgaaagtcaacgagatgacacatctgttatcacttactgtacaagcaaagcactattttcactgctactggaatggtctgtgtttaatttatagttgccaatcctgtagcgac-------------------------
ERR4451134.487 M00758:969:CG8RD:1:1102:16023:2116/1 ---------------------ctcctcataataattagtaatatctctgctatagtaacctgaaagtcaacgagatgaaacatctgttgtcacttactgtacaagcaaagcaatattgtcactgctactggaatggtctgtgtttaatttatagttgccaatcctgtagcg---------------------------
ERR4451134.566 M00758:969:CG8RD:1:1102:15979:2131/1 ------------------------ctcataataattagtaatatctctgctatagtaacctgaaagtcaacgagatgaaacatctgttgtcacttactgtacaagcaaagcaatattgtcactgctactggaatggtctgtgtttaatttatagttgccaatcctgtagcgact------------------------
$ seqkit fx2tab temp.aln|cut -f2|gawk -F '' '{for(i=1;i<=length;i++)a[i][$i]++}END{for(i in a){max=0;for(j in a[i])if(a[i][j]>max){max=a[i][j];o=j}printf"%s",o};print""}'|tee consensus
-----------------------cctcataataattagtaatatctctgctatagtaacctgaaagtcaacgagatgaaacatctgttgtcacttactgtacaagcaaagcaatattgtcactgctactggaatggtctgtgtttaatttatagttgccaatcctgtagcgac-------------------------
$ cons temp.aln -outseq /dev/stdout|seqkit seq -w0 # `cons` is from EMBOSS
Create a consensus sequence from a multiple alignment
>EMBOSS_001
nnnnnnnnnnnnnnnnnnnnnnnnCTCATAATAATTAGTAATATCTCTGCTATAGTAACCTGAAAGTCAACGAGATGAAACATCTGTTGTCACTTACTGTACAAGCAAAGCAATATTGTCACTGCTACTGGAATGGTCTGTGTTTAATTTATAGTTGCCAATCCTGTAGCGACTnnnnnnnnnnnnnnnnnnnnnnnn
$ Rscript -e 'writeLines(Biostrings::consensusString(Biostrings::readAAStringSet("temp.aln")))'
------------------------ctcataataattagtaatatctctgctatagtaacctgaaagtcaacgagatgaaacatctgttgtcacttactgtacaagcaaagcaatattgtcactgctactggaatggtctgtgtttaatttatagttgccaatcctgtagcgact------------------------
$ awk 'NR==1{split($0,a,"");l=length;next}{split($0,b,"");n=0;for(i=1;i<=l;i++)if(a[i]!="-"&&b[i]!="-"&&a[i]!=b[i])n++;print n}' consensus <(seqkit seq -s temp.aln)|sort|uniq -c # 415 reads have 0 nucleotide changes from consensus, 61 reads have 1 change, etc.
414 0
61 1
11 2
5 3
3 4
2 5
2 6
1 7
1 8
This compares the sequence whose ID is specified as an argument against every sequence in the FASTA file from STDIN. It ignores positions where either sequence has a gap (because often for example some sequences are missing parts from the 5' or 3' UTRs which can artificially inflate their distance to other sequences if gaps are not ignored):
$ pid1(){ x=$(cat);awk 'NR==1{split($0,a,"");l=length;next}{split($0,b,"");d=0;n=0;for(i=1;i<=l;i++)if(a[i]!="-"&&b[i]!="-"){n++;if(a[i]!=b[i])d++}print 100*(1-d/n)}' <(seqkit grep -p "$1"<<<"$x"|seqkit seq -s;seqkit seq -s<<<"$x");}
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=NC_045512,NC_004718,MN996532,MG772933,MZ937000,MZ190137,KY352407'|mafft --quiet --reorder ->temp.fa
$ pid1 NC_006577.1 <temp.fa|paste - <(seqkit seq -n temp.fa|sed 's/, complete genome//')
100 NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1
96.967 MZ937000.1 Bat coronavirus isolate BANAL-20-52/Laos/2020
96.1983 MN996532.2 Bat coronavirus RaTG13
88.0544 MG772933.1 Bat SARS-like coronavirus isolate bat-SL-CoVZC45
79.8987 NC_004718.3 SARS coronavirus Tor2
76.5423 MZ190137.1 Bat SARS-like coronavirus Khosta-1 strain BtCoV/Khosta-1/Rh/Russia/2020
76.6389 KY352407.1 Severe acute respiratory syndrome-related coronavirus strain BtKY72
This compares each 2-combination of sequences and sorts the results based on the percentage identity:
$ seqkit fx2tab temp.fa|sed 's/, complete genome//;s/.* //'|awk -F\\t '{split($2,x,"");l=length($2);for(i=1;i<=l;i++)a[NR][i]=x[i];name[NR]=$1;seqs=NR}END{for(i=1;i<=seqs-1;i++)for(j=i+1;j<=seqs;j++){d=0;n=0;for(k=1;k<=l;k++)if(a[i][k]!="-"&&a[j][k]!="-"){n++;if(a[i][k]!=a[j][k])d++}print 100*(1-d/n)"\t"name[i]"\t"name[j]}}'|sort -rn|column -ts$'\t'
96.967 Wuhan-Hu-1 BANAL-20-52/Laos/2020
96.1983 Wuhan-Hu-1 RaTG13
96.0374 BANAL-20-52/Laos/2020 RaTG13
88.0656 BANAL-20-52/Laos/2020 bat-SL-CoVZC45
88.0544 Wuhan-Hu-1 bat-SL-CoVZC45
87.9844 RaTG13 bat-SL-CoVZC45
81.7179 BtCoV/Khosta-1/Rh/Russia/2020 BtKY72
81.2542 bat-SL-CoVZC45 Tor2
79.9074 BANAL-20-52/Laos/2020 Tor2
79.8987 Wuhan-Hu-1 Tor2
79.7034 RaTG13 Tor2
78.8248 Tor2 BtCoV/Khosta-1/Rh/Russia/2020
77.9839 Tor2 BtKY72
77.1394 bat-SL-CoVZC45 BtCoV/Khosta-1/Rh/Russia/2020
76.8069 bat-SL-CoVZC45 BtKY72
76.6909 BANAL-20-52/Laos/2020 BtKY72
76.6389 Wuhan-Hu-1 BtKY72
76.6371 BANAL-20-52/Laos/2020 BtCoV/Khosta-1/Rh/Russia/2020
76.5523 RaTG13 BtKY72
76.5423 Wuhan-Hu-1 BtCoV/Khosta-1/Rh/Russia/2020
76.4696 RaTG13 BtCoV/Khosta-1/Rh/Russia/2020
#include <iostream>
#include <vector>
#include <string>
#include <cctype>
using namespace std;
int main(int argc,char**argv){
string id,buf;
vector<string>seq,name;
for(string line;getline(cin,line);){
if(line.empty())continue;
if(line[0]=='>'){
if(!id.empty()){
name.push_back(id);
transform(buf.begin(),buf.end(),buf.begin(),::tolower);
seq.push_back(buf);
}
id=line.substr(1);
buf.clear();
}else buf+=line;
}
if(!id.empty()){
name.push_back(id);
transform(buf.begin(),buf.end(),buf.begin(),::tolower);
seq.push_back(buf);
}
int seqs=seq.size(),positions=seq[0].size();
vector<vector<int> >diffs(seqs,vector<int>(seqs));
vector<vector<int> >totals(seqs,vector<int>(seqs));
int ind=0;
for(int i=0;i<seqs-1;i++){
for(int j=i;j<seqs;j++){
int diff=0,total=0;
for(int k=0;k<positions;k++)
if(seq[i][k]!='-'&&seq[j][k]!='-'){
total++;
if(seq[i][k]!=seq[j][k])diff++;
}
diffs[i][j]=diff;
totals[i][j]=total;
}
}
for(int i=0;i<seqs;i++)cout<<"\t"<<name[i];
cout<<endl;
for(int i=0;i<seqs;i++){
cout<<name[i];
for(int j=0;j<seqs;j++){
int x=i<j?i:j,y=i<j?j:i;
int diff=diffs[x][y],total=i==j?1:totals[x][y];
printf("\t%.4f",100*(1-(float)diff/total));
}
cout<<endl;
}
}
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=NC_019843,NC_039207,NC_014470,NC_006577'|mafft --quiet --reorder ->temp.fa
$ tab(){ awk '{if(NF>m)m=NF;for(i=1;i<=NF;i++){a[NR][i]=$i;l=length($i);if(l>b[i])b[i]=l}}END{for(h in a){for(i=1;i<=m;i++)printf(i==m?"%s\n":"%-"(b[i]+n)"s",a[h][i])}}' "${1+FS=$1}" "n=${2-1}";}
$ g++ -O3 -o pid pid.cpp
$ sed 's/, complete genome//;s/>.* />/' temp.fa|./pid|tab \\t
Wuhan-Hu-1 BANAL-20-52/Laos/2020 RaTG13 bat-SL-CoVZC45 bat-SL-CoVZXC21 Tor2
Wuhan-Hu-1 100.0000 96.9669 96.1983 88.0342 88.0136 79.9149
BANAL-20-52/Laos/2020 96.9669 100.0000 96.0374 88.0455 88.0296 79.9270
RaTG13 96.1983 96.0374 100.0000 87.9676 87.9973 79.7196
bat-SL-CoVZC45 88.0342 88.0455 87.9676 100.0000 97.4706 81.2809
bat-SL-CoVZXC21 88.0136 88.0296 87.9973 97.4706 100.0000 81.3978
Tor2 79.9149 79.9270 79.7196 81.2809 81.3978 100.0000
The code ignores positions where either sequence has a gap, but if
you also want to ignore N bases and degenerate bases, you can replace
them with gaps: seqkit replace -isp'[^acgt]' -r-.
# download a FASTA file for the thousand closest matches to the spike protein of Wuhan-Hu-1 on BLAST with the taxid of SARS2 excluded
curl -Lso spike1k.fa 'https://drive.google.com/uc?export=download&id=1riMniCLMe4Aqg-hj4O6LXQaqvx2gPnHx'
curl -Lso spike1k.tsv 'https://drive.google.com/uc?export=download&id=1o79qDLcrbxAppnK15t7tjdn7vKU2H3dT'
# omit sequences with too many X letters or gaps and realign the sequences
seqkit seq -s spike1k.fa|tr -d X-|awk '{print length}'|paste - <(seqkit seq -ni spike1k.fa)|awk '$1>=1200'|cut -f2|seqkit grep -f- spike1k.fa|mafft --reorder ->spike1k.aln
# `-a` counts all letters and not just ACGT
snp-dists -a spike1k.aln>spike1k.dist
t=read.table("spike1k.dist",sep="\t",r=1,head=T,quote="",comment.char="",check.names=F)
t2=read.table("spike1k.tsv",sep="\t",r=1,head=T,quote="",comment.char="",check.names=F)
name=apply(t2[rownames(t),c(11,5,6,7)],1,paste,collapse="|")
pick=!grepl("Synthetic|Recombinant|Cloning|Expression",name)
t=t[pick,pick]
name=name[pick]
colnames(t)=name
pick2=grepl("BANAL",name)
r=apply(t[pick2,],1,\(x){h=sort(x)[2:5];paste(" ",h,names(h),collapse="\n")})
writeLines(paste0(name[pick2],"\n",r))
From the output you can see that BANAL-52 is similar to SARS2, BANAL-103 and -236 are similar to Chinese pangolin viruses, and BANAL-116 and -247 are similar to the Thai bat coronavirus RacCS203:
Bat coronavirus BANAL-20-52/Laos/2020|2021-09-19|2020-07-05|Laos 20 SARS coronavirus 2 Wuhan-Hu-1|2020-01-13|2019-12|China 28 Bat coronavirus RaTG13|2020-01-29|2013-07-24|China 29 Bat SARS-like virus BtSY2|2023-01-24|2018|China: Yunnan 91 Pangolin coronavirus GX_P2V|2022-02-01|| Bat coronavirus BANAL-20-103/Laos/2020|2021-09-19|2020-07-07|Laos 5 Bat coronavirus BANAL-20-236/Laos/2020|2021-09-19|2020-07-10|Laos 35 Pangolin coronavirus cDNA8-S|2020-08-01|2019|China 35 Pangolin coronavirus cDNA18-S|2020-08-01|2019|China 36 Pangolin coronavirus cDNA16-S|2020-08-01|2019|China Bat coronavirus BANAL-20-236/Laos/2020|2021-09-19|2020-07-10|Laos 5 Bat coronavirus BANAL-20-103/Laos/2020|2021-09-19|2020-07-07|Laos 37 Pangolin coronavirus cDNA8-S|2020-08-01|2019|China 37 Pangolin coronavirus cDNA18-S|2020-08-01|2019|China 38 Pangolin coronavirus cDNA16-S|2020-08-01|2019|China Bat coronavirus BANAL-20-247/Laos/2020|2021-09-19|2020-07-10|Laos 4 Bat coronavirus BANAL-20-116/Laos/2020|2021-09-19|2020-07-07|Laos 25 Bat coronavirus RacCS203|2021-01-13|2020-06-19|Thailand 210 Sarbecovirus sp. HN2021C|2021-09-22|2021-02|China: Hunan 215 Bat SARS-like coronavirus Rf4092|2017-11-01|2012-09-18|China Bat coronavirus BANAL-20-116/Laos/2020|2021-09-19|2020-07-07|Laos 4 Bat coronavirus BANAL-20-247/Laos/2020|2021-09-19|2020-07-10|Laos 26 Bat coronavirus RacCS203|2021-01-13|2020-06-19|Thailand 211 Sarbecovirus sp. HN2021C|2021-09-22|2021-02|China: Hunan 216 Bat SARS-like coronavirus Rf4092|2017-11-01|2012-09-18|China
library(ggplot2)
t=as.matrix(Biostrings::readAAStringSet("https://pastebin.com/raw/h0nHf55b"))
targ="ZC45"
step=100
window=500
xbreak=2000
ybreak=10
t1=t[rownames(t)!=targ,]
t2=t[targ,]
ncol=ncol(t)
starts=seq(1,ncol,step)
sim=apply(t1,1,\(seq)sapply(starts,\(i){
start=max(1,i-window%/%2)
end=min(ncol,i+window%/%2)
mean(seq[start:end]==t2[start:end])
}))
long=data.frame(V1=starts[row(sim)],V2=colnames(sim)[col(sim)],V3=100*c(sim))
group=factor(long$V2,levels=unique(long$V2))
hue=c(0,210,100,60,300,160,30,250)+15
color=c("black",hcl(hue,60,70),"gray50",hcl(hue,50,40))
rmu=\(x,y)x+(y-x%%y)%%y
xend=rmu(max(long$V1-1),xbreak)+1
ystart=rmu(min(long$V3),-ybreak)
yend=rmu(max(long$V3),ybreak)
ggplot(long,aes(x=V1,y=V3))+
geom_line(aes(color=group),linewidth=.3)+
geom_vline(xintercept=ncol,color="gray90",linewidth=.3,linetype="dashed")+
ggtitle(paste0("Similarity to ",targ," (step ",step,", window ",window,")"))+
scale_x_continuous(limits=c(1,xend),breaks=seq(1,xend,xbreak),expand=expansion(mult=c(.005,.005)))+
scale_y_continuous(limits=c(ystart,yend),breaks=seq(ystart,yend,ybreak),expand=expansion(mult=c(.012,.012)))+
scale_color_manual(values=color)+
guides(color=guide_legend(ncol=4,byrow=T))+
theme(
axis.text=element_text(size=6,color="black"),
axis.text.x=element_text(angle=90,vjust=.5,hjust=1),
axis.ticks=element_blank(),
axis.ticks.length=unit(0,"cm"),
axis.title=element_blank(),
legend.background=element_blank(),
legend.box.margin=margin(0),
legend.box.spacing=unit(0,"in"),
legend.key=element_rect(fill=NA),
legend.margin=margin(-4,0,-1.5,0),
legend.position="top",
## legend.position=c(.7,.2),
legend.spacing.x=unit(.03,"in"),
legend.spacing.y=unit(0,"in"),
legend.text=element_text(size=7,vjust=.5),
legend.title=element_blank(),
panel.background=element_rect(fill="white"),
panel.grid.major=element_line(linewidth=.2,color="gray90"),
panel.grid.minor=element_blank(),
plot.background=element_rect(fill="white"),
plot.title=element_text(size=9,color="black")
)
ggsave("1.png",width=4.5,height=3)
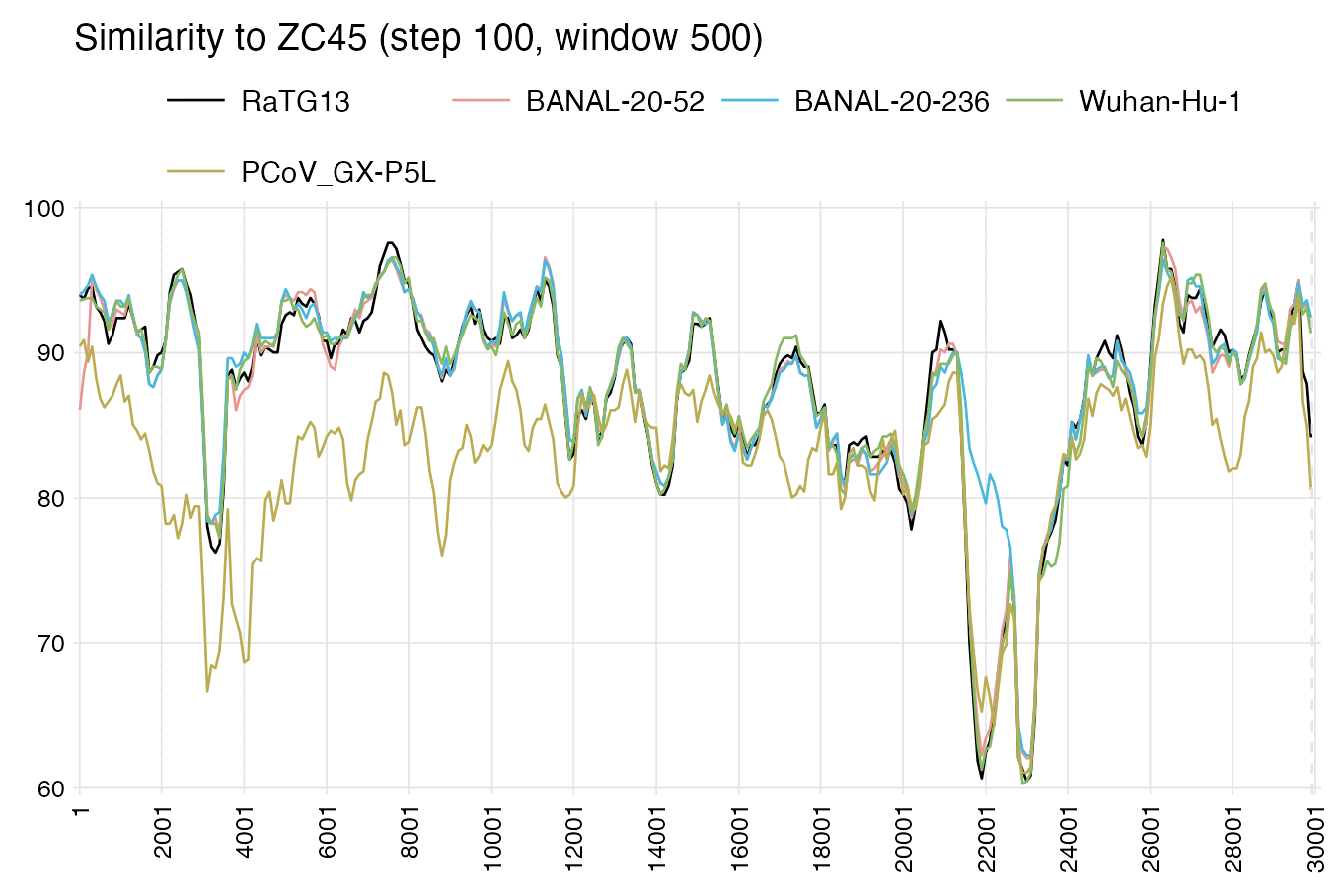
There's also a JavaScript utility for creating simplots: http://babarlelephant.free-hoster.net/simplotfirstN.html.
library(ggplot2)
t=as.matrix(Biostrings::readAAStringSet("input.fa"))
gene=read.csv(head=F,text="ORF1a,266,13468
ORF1b,13468,21555
S,21563,25384
ORF3a,25393,26220
E,26245,26472
M,26523,27191
ORF6,27202,27387
ORF7a,27394,27759
ORF8,27894,28259
N,28274,29533
ORF10,29558,29674")
ref=t[grepl("Wuhan-Hu-1",rownames(t)),]
pos=seq_along(ref)-cumsum(ref=="-")
gene[,2]=sapply(gene[,2],\(x)which(pos==x)[1])
gene[,3]=sapply(gene[,3],\(x)which(pos==x)[1])
genebars=c(gene[,2],gene[nrow(gene),2])
rownames(t)=sub("(,| ORF| surface ).*","",rownames(t))
rownames(t)=sub("[^ ]* ","",rownames(t))
rownames(t)=sub(" isolate "," ",rownames(t))
rownames(t)=sub("Severe acute respiratory syndrome coronavirus 2 ","",rownames(t))
targ=rownames(t)[grep("Tor2",rownames(t))]
window=500
step=100
t1=t[rownames(t)!=targ,]
t2=t[targ,]
ncol=ncol(t)
starts=seq(1,ncol,step)
sim=apply(t1,1,\(seq)sapply(starts,\(i){
start=max(1,i-window%/%2)
end=min(ncol,i+window%/%2)
mean((seq[start:end]==t2[start:end])[seq[start:end]!="-"&t2[start:end]!="-"])
}))
xy=data.frame(x=starts[row(sim)],y=100*c(sim),group=colnames(sim)[col(sim)])
xy$group=factor(xy$group,unique(xy$group))
hue=c(0,210,100,60,300,160,30,250)+15
color=c("black",hcl(hue,80,70),"gray50",hcl(hue,60,40))
color=setNames(color[1:nrow(t)],rownames(t))[levels(xy$group)]
candidates=c(sapply(c(1,2,5),\(x)x*10^c(-10:10)))
xbreak=candidates[which.min(abs(candidates-max(xy$x)/10))]
ybreak=candidates[which.min(abs(candidates-(max(xy$y)-min(xy$y))/7))]
xend=max(xy$x)
ystart=ybreak*floor(min(xy$y,na.rm=T)/ybreak)
yend=ybreak*ceiling(max(xy$y,na.rm=T)/ybreak)
legend=data.frame(x=.02*xend,y=.02*(yend-ystart)+rev(seq(ystart+(yend-ystart)*.05,by=(yend-ystart)/24,length.out=nlevels(xy$group))),label=levels(xy$group))
label=data.frame(label=gene[,1],x=rowMeans(gene[,-1]),y=ystart+(yend-ystart)*(c(1,1,1,1,2,3,1,2,3,1,2)-.2)/32)
ggplot(xy)+
# geom_rect(data=gene[c(T,F),],aes(xmin=V2,xmax=V3,ymin=ystart,ymax=yend),fill="gray90")+
geom_vline(xintercept=genebars,color="gray80",linewidth=.25)+
geom_vline(xintercept=c(0,xend),color="gray80",linewidth=.25)+
geom_line(aes(x,y,color=group),linewidth=.25)+
geom_label(data=legend,aes(x,y,label=label),color=color,size=2.4,hjust=0,fill=alpha("white",.7),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0)+
geom_label(data=label,aes(x=x,y=y,label=label),fill=alpha("white",.7),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0,size=1.8,hjust=.5)+
ggtitle(paste0("Similarity to ",targ," (step ",step,", window ",window,")"))+
coord_cartesian(clip="off")+
scale_x_continuous(limits=c(0,xend),breaks=seq(0,xend,xbreak),expand=c(0,0),labels=\(x)ifelse(x>1e3,paste0(x/1e3,"k"),x))+
# scale_x_continuous(limits=c(0,xend),breaks=seq(0,xend,xbreak),expand=c(0,0))+
scale_y_continuous(limits=c(ystart,yend),breaks=seq(ystart,yend,ybreak),expand=c(0,0))+
scale_color_manual(values=color)+
theme(
axis.text=element_text(size=6,color="black"),
axis.ticks=element_line(linewidth=.25,color="gray80"),
axis.ticks.length=unit(.2,"lines"),
axis.title=element_blank(),
legend.position="none",
panel.background=element_rect(fill="white"),
panel.grid.major=element_blank(),
panel.grid.major.y=element_line(linewidth=.25,color="gray80"),
plot.background=element_rect(fill="white"),
plot.margin=margin(.5,.6,.5,.6,"lines"),
plot.title=element_text(size=8,color="black")
)
ggsave("1.png",width=5,height=3.5)
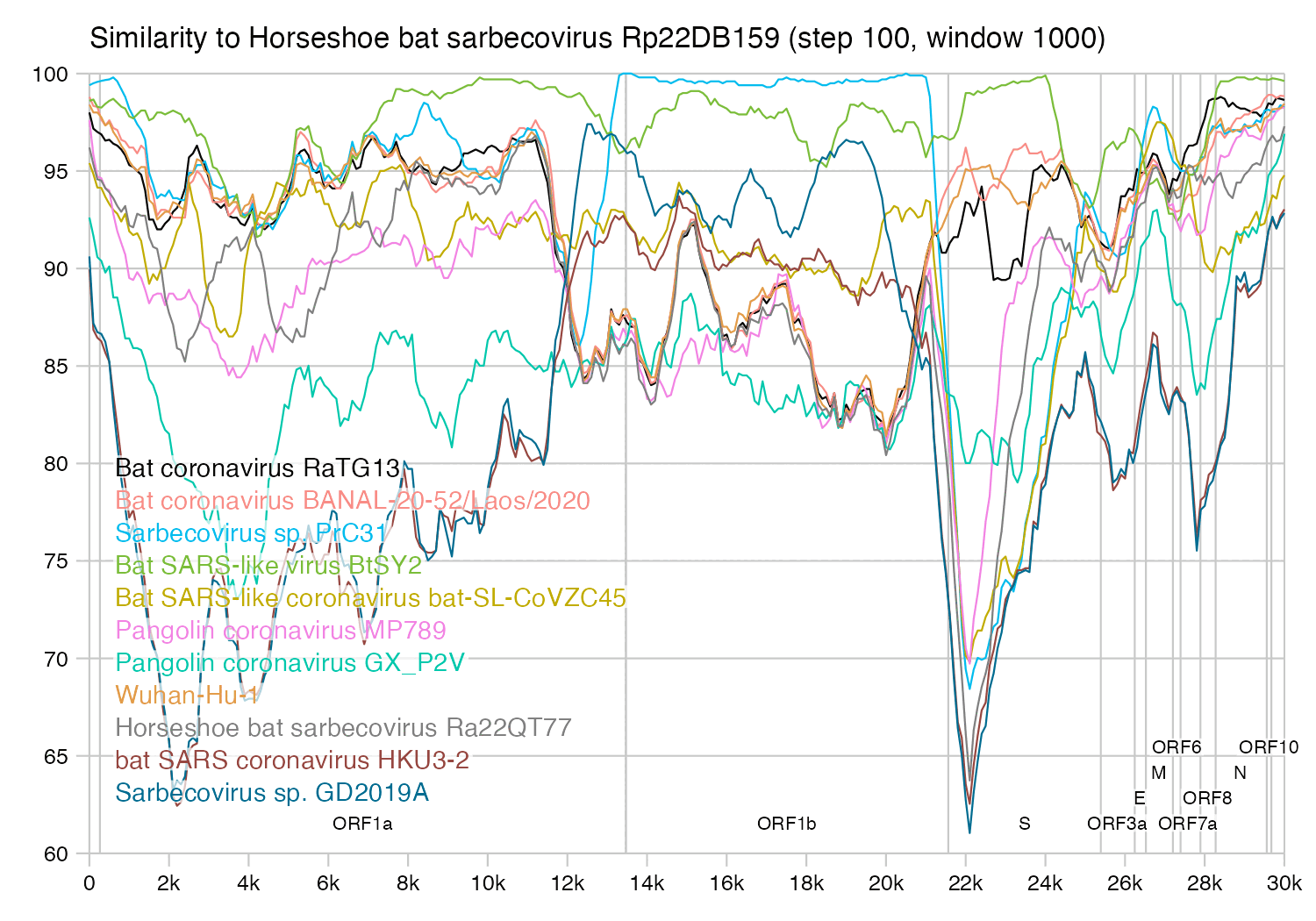
Rp22DB159 is one of the new Vietnamese sequences from Hassanin et al. 2023. It's a recombinant virus where some regions are similar to SARS2-like viruses and other regions are similar to SARS1-like viruses, like in ZC45, BtSY2, PrC31, and YN2021. The plot above shows that in Rp22DB159 the S1 is similar to BtSY2 but the ORF1b is similar to PrC31.
curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh|sh
esearch -db nuccore -query '(viruses[filter] AND sarbecovirus NOT txid2697049) OR NC_045512.2 OR OM240725 OR MZ328294 OR (Bat coronavirus isolate BANAL-20)'|efetch -format fasta|seqkit seq -m 25000 >sarbe0.fa
seqkit grep -nrvp '^[^ ]* [^ ]* [^ ]*:|Patent|MODIFIED SMALL INTERFERING|Modified Microbial|A sensitive and specific test|NOVEL STRAIN|IMAGEABLE|isolate H_SC_|AY274119.3|Tor2 isolate' sarbe0.fa|mafft --thread 4 --reorder ->sarbe.fa
curl https://pastebin.com/raw/pDwYNf1r>pid.cpp;g++ pid.cpp -O3 -o pid
# ignore sequences with 1000 or more N letters or degenerate bases
seqkit seq -s sarbe.fa|tr -d ACGT-|paste - <(seqkit seq -ni sarbe.fa)|awk 'length($1)>1000{print$2}'>sarbe.ignore
# matrix sort grep (sort by first column matching regex and print the value of the column with rownames)
masog()(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~x)break;next}{print$i,$1}' x="$1" "${2--}"|sort -n)
# ignore other SARS1 sequences except Tor2
masog Tor2, sarbe.pid|awk '$1<100&&$1>98{print$2}'>>sarbe.ignore
# `pid` ignores positions where either sequence has a gap, so N letters and degenerate bases are replaced with gaps
seqkit grep -vf sarbe.ignore sarbe.fa|seqkit replace -isp '[^ACGT-]' -r -|./pid>sarbe.pid
I uploaded the output files here: f/sarbe.fa.xz, f/sarbe.pid.
The sequence of the Guangdong pangolin virus GD/P2S is available from GISAID but not GenBank. The sequence has about 30% N bases, but if you ignore positions with an N letter, degenerate base, or a gap, then GD/P2S has about 99.6-99.7% identity with MP789 and GD/P79-9 and about 99.1% identity with GD/P44-9. Therefore in order to fill in the N letters and degenerate bases, you can replace them with the consensus sequence of other pangolin viruses:
$ curl https://pastebin.com/raw/AuXrLcLx|tr -d \\r>gdp2s.fa
$ seqkit subseq -r1:200 gdp2s.fa
>hCoV-19/pangolin/Guangdong/P2S/2019
TTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTAA
TAACTAATTACTGKCGTTGACAGGACACGAGTAACTCGTCTATCTTCTGCNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGCATACCTAGGTTTCGTCCGGGTGTGACCG
AAAGGTAAGATGGAGAGCCT
$ curl https://sars2.net/f/sarbe.fa.xz|xz -dc>sarbe.fa
$ seqkit grep -nrp 'GD/P44-9|GD/P44-9|MT121216' sarbe.fa|cat gdp2s.fa -|mafft --thread 4 ->pang.fa
[...]
$ seqkit replace -isp '[^acgtn-]' -r n pang.fa|seqkit seq -s|gawk -F '' '{for(i=1;i<=NF;i++)if($i!="n")a[i][$i]++}END{for(i in a){max=0;for(j in a[i])if(a[i][j]>max){max=a[i][j];o=j}printf"%s",o};print""}'|cat - <(seqkit seq -s pang.fa|sed 1q)|gawk -F '' 'NR==1{split($0,a,"");next}{for(i=1;i<=NF;i++)printf"%s",$i=="n"?a[i]:$i}'|(echo \>filled_in;tr -d -)|seqkit subseq -r1:200
>filled_in
ttaaaatctgtgtggctgtcactcggctgcatgcttagtgcactcacgcagtataattaa
taactaattactgkcgttgacaggacacgagtaactcgtctatcttctgctgcttagact
cggtggagatattgttagaactaatatatcgcatacctaggtttcgtccgggtgtgaccg
aaaggtaagatggagagcct
This downloads metadata about a global subsample of about 14,000 SARS2 sequences from NextStrain:
curl https://data.nextstrain.org/files/ncov/open/{africa,asia,europe,north-america,oceania,south-america}/metadata.tsv.xz|gzip -dc|awk 'NR==1||$1!="strain"'>continent.tsv
library(ggplot2)
meta=read.table("continent.tsv",header=T,sep="\t",quote="")
pick=meta$divergence<200&meta$length>29e3&meta$Nextstrain_clade!=""&meta$Nextstrain_clade!="recombinant"
pick=pick&!(grepl("Omicron",meta$Nextstrain_clade)&as.Date(meta$date)<as.Date("2021-10-1"))
pick=pick&!(grepl("19B",meta$Nextstrain_clade)&as.Date(meta$date)>as.Date("2021-10-1"))
pick=pick&!(grepl("19A",meta$Nextstrain_clade)&as.Date(meta$date)>as.Date("2021-10-1"))
pick=pick&!(grepl("20A",meta$Nextstrain_clade)&as.Date(meta$date)>as.Date("2021-08-1"))
pick=pick&!(grepl("20G",meta$Nextstrain_clade)&as.Date(meta$date)<as.Date("2020-07-1"))
pick=pick&!(grepl("20B",meta$Nextstrain_clade)&as.Date(meta$date)>as.Date("2021-10-1"))
pick=pick&!(grepl("21H",meta$Nextstrain_clade)&as.Date(meta$date)<as.Date("2020-10-1"))
pick=pick&!(grepl("21J",meta$Nextstrain_clade)&as.Date(meta$date)<as.Date("2020-10-1"))
pick=pick&!(grepl("Gamma",meta$Nextstrain_clade)&as.Date(meta$date)<as.Date("2020-12-1"))
meta=meta[pick,]
month=tapply(meta$divergence,sub("-..$","",meta$date),mean)
xy=data.frame(x=as.Date(paste0(names(month),"-1")),y=month)
t=aggregate(meta$divergence,list(sub("-..$","",meta$date),meta$Nextstrain_clade),mean)
count=aggregate(meta$divergence,list(sub("-..$","",meta$date),meta$Nextstrain_clade),length)
t=t[count[,3]>1,]
colnames(t)=paste0("V",1:3)
t[,1]=as.Date(paste0(t[,1],"-1"))
group=factor(t[,2],unique(t[,2]))
hue=c(0,30,60,90,150,210,250,310)+15
color=rep(c(hcl(hue,60,80),hcl(hue,100,40)),3)
xstart=as.Date("2019-12-1")
xend=as.Date("2023-08-01")
xbreak=seq(xstart,xend,"2 months")
ystart=0
yend=120
ybreak=seq(ystart,yend,10)
ggplot(t,aes(x=V1,y=V3,color=V2,fill=V2,linetype=V2))+
geom_vline(xintercept=c(xstart,xend),color="gray75",linewidth=.35,lineend="square")+
geom_hline(yintercept=c(ystart,yend),color="gray75",linewidth=.35,lineend="square")+
geom_point(size=.5)+
geom_line(linewidth=.5)+
labs(title="Monthly average divergence from Wuhan-Hu-1 by strain among 14,358 samples from NextStrain",x=NULL,y="Nucleotide changes from Wuhan-Hu-1")+
scale_x_date(limits=c(xstart,xend),breaks=xbreak,expand=expansion(0),date_labels="%b %y")+
scale_y_continuous(limits=c(ystart,yend),breaks=ybreak,expand=expansion(0))+
scale_color_manual(values=color)+
scale_fill_manual(values=color)+
scale_linetype_manual(values=rep(c(1,2,5),each=16))+
coord_cartesian(clip="off")+
guides(color=guide_legend(nrow=9))+
theme(
axis.text.x=element_text(angle=90,vjust=.5,hjust=1),
axis.text=element_text(size=7,color="black"),
axis.ticks=element_line(linewidth=.35,color="gray75"),
axis.ticks.length=unit(.2,"lines"),
axis.title=element_text(size=8),
legend.background=element_blank(),
legend.box.just="center",
legend.box.margin=margin(0,unit="cm"),
legend.box.spacing=unit(0,"in"),
legend.direction="vertical",
legend.justification="left",
legend.key=element_rect(fill=NA),
legend.position=c(.0,.73),
legend.text=element_text(size=7,vjust=.5),
legend.title=element_blank(),
panel.grid=element_blank(),
panel.background=element_rect(fill="white"),
plot.margin=margin(.4,.6,.4,.6,"lines"),
plot.title=element_text(size=9)
)
ggsave("1.png",width=6,height=4.5)
Even though the script filtered out many samples with incorrect collection date, some samples with an incorrect collection date remained, like one Mu sample from July 2020. But you can still see that for example in December 2020, the line for Alpha starts a lot higher than the average height of the lines for other variants in November:
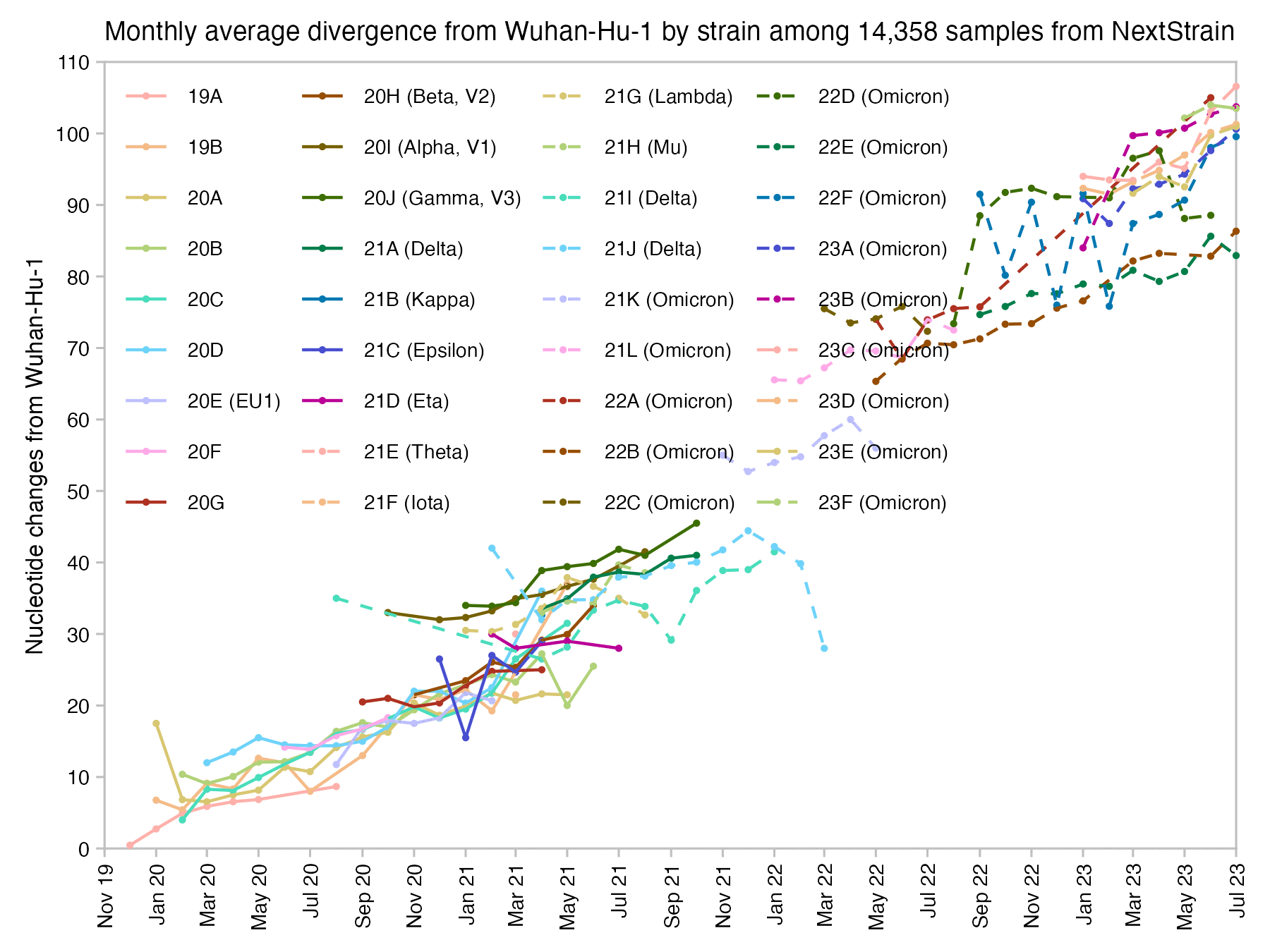
Wikipedia says that almost all known RNA viruses are contained within the realm of Riboviria: "As of May 2020, all known RNA viruses encoding an RNA-directed RNA polymerase are believed to form a monophyletic group, known as the realm Riboviria.[4] The majority of such RNA viruses fall into the kingdom Orthornavirae and the rest have a positioning not yet defined.[5] The realm does not contain all RNA viruses: Deltavirus, Asunviroidae, and Pospiviroidae are taxa of RNA viruses that were mistakenly included in 2019,[a] but corrected in 2020.[6]" The genus Deltavirus consists solely of hepatitis delta which has an extremely short genome, and Asunviroidae and Pospiviroidae are families of viroids that have short genomes.
I wanted to search which RNA viruses had the longest genomes, so I
searched the NCBI's nucleotide database for all reference sequences
classified under the realm of Riboviria.
[https://www.ncbi.nlm.nih.gov/nuccore/?term=%22Riboviria%22%5BOrganism%5D+AND+refseq%5Bfilter%5D]
You can download a FASTA file of the results from the web GUI by
clicking "Send to", setting the format to
FASTA, and clicking "Create File". Or
alternatively you can run this:
esearch -db nuccore -query '"Riboviria"[Organism] AND refseq[filter]'|efetch -format fasta.
There was a total of 9,116 results. The longest sequence among the results was 41,178 bases long and titled "Planarian secretory cell nidovirus isolate UIUC". It was published in a paper from 2018 titled "A planarian nidovirus expands the limits of RNA genome size". The longest coronavirus was "Beluga whale coronavirus SW1" which was 31,686 bases long and came on 8th place:
$ curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh|sh
$ esearch -db nuccore -query '"Riboviria"[Organism] AND refseq[filter]'|efetch -format fasta>riboviridae.fa
$ seqkit fx2tab riboviridae.fa|awk -F\\t '{print length($2),$1}'|sort -nr|head
41178 NC_040361.1 Planarian secretory cell nidovirus isolate UIUC, complete genome
36652 NC_076697.1 MAG: Pacific salmon nidovirus isolate H14 ORF1a polyprotein (QKQ71_gp1), ORF1b polyprotein (QKQ71_gp2), LAP1C-like protein (QKQ71_gp3), spike protein (QKQ71_gp4), envelope (QKQ71_gp5), membrane protein (QKQ71_gp6), and nucleoprotein (QKQ71_gp7) genes, complete cds
36144 NC_076911.1 Veiled chameleon serpentovirus B, complete sequence
35906 NC_040711.1 Aplysia californica nido-like virus isolate EK, complete genome
33452 NC_024709.1 Ball python nidovirus strain 07-53, complete genome
32753 NC_076657.1 Morelia viridis nidovirus isolate BH171/14-7, complete genome
32399 NC_035465.1 Morelia viridis nidovirus strain S14-1323_MVNV, complete genome
31686 NC_010646.1 Beluga Whale coronavirus SW1, complete genome
31537 NC_076912.1 Veiled chameleon serpentovirus A, complete sequence
31526 AC_000192.1 Murine hepatitis virus strain JHM, complete genome
Downloading reads from SRA with fastq-dump is so slow
that people have developed tools like fasterq-dump and
parallel-fastq-dump which run multiple
fastq-dump processes in parallel. However
fasterq-dump doesn't support downloading gzipped reads. An
easy way to download gzipped reads fast is to just use ENA instead:
$ enal()(printf %s\\n "${@-$(cat)}"|while read x;do curl -s "https://www.ebi.ac.uk/ena/portal/api/filereport?accession=$x&result=read_run&fields=fastq_ftp"|sed 1d|cut -f2|tr \; \\n|sed s,^,ftp://,;done) # ENA list
$ enad()(enal "$@"|xargs wget) # ENA download
$ enal SRR10971381
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR109/081/SRR10971381/SRR10971381_1.fastq.gz
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR109/081/SRR10971381/SRR10971381_2.fastq.gz
$ curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh|sh [...] $ esearch -db sra -query 'pangolin OR manis'|efetch -format runinfo>pangolin.runinfo $ sed '1n;/Manis/!d' pangolin.runinfo|head -n3 Run,ReleaseDate,LoadDate,spots,bases,spots_with_mates,avgLength,size_MB,AssemblyName,download_path,Experiment,LibraryName,LibraryStrategy,LibrarySelection,LibrarySource,LibraryLayout,InsertSize,InsertDev,Platform,Model,SRAStudy,BioProject,Study_Pubmed_id,ProjectID,Sample,BioSample,SampleType,TaxID,ScientificName,SampleName,g1k_pop_code,source,g1k_analysis_group,Subject_ID,Sex,Disease,Tumor,Affection_Status,Analyte_Type,Histological_Type,Body_Site,CenterName,Submission,dbgap_study_accession,Consent,RunHash,ReadHash SRR24150370,2023-04-13 00:39:14,2023-04-12 23:17:46,1320057,356415390,1320057,270,106,,https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos4/sra-pub-zq-3/SRR024/24150/SRR24150370/SRR24150370.lite.1,SRX19948497,HC-HKU06,RAD-Seq,Restriction Digest,GENOMIC,PAIRED,0,0,ILLUMINA,Illumina NovaSeq 6000,SRP432345,PRJNA954554,,954554,SRS17296698,SAMN34161674,simple,9974,Manis javanica,HKU06,,,,,female,,no,,,,,THE UNIVERSITY OF HONG KONG,SRA1619804,,public,3C0FCC08646788EEAD35C7134007B7B5,26E77FA1DF1CC34214421679C1BED294 SRR24150359,2023-04-13 00:39:14,2023-04-12 23:17:44,837476,226118520,837476,270,68,,https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos4/sra-pub-zq-3/SRR024/24150/SRR24150359/SRR24150359.lite.1,SRX19948508,LC-HKU07,RAD-Seq,Restriction Digest,GENOMIC,PAIRED,0,0,ILLUMINA,Illumina NovaSeq 6000,SRP432345,PRJNA954554,,954554,SRS17296709,SAMN34161675,simple,9974,Manis javanica,HKU07,,,,,male,,no,,,,,THE UNIVERSITY OF HONG KONG,SRA1619804,,public,B34A9AC17DAB6A628733E087D7F5D80B,F1E59AB9CC3872A2B867863FFDE81792
This downloads FASTQ files for all runs in a BioProject:
esearch -db sra -query PRJNA605983|efetch -format runinfo|sed 1d|cut -d, -f1|parallel fastq-dump --gzip --split-3
This code finds the longest direct repeat within a sequence, and it prints the sequence of the repeat along with the length, starting position, and ending position of each instance of the repeat:
$ printf %s\\n \>sv40_promoter GGTGTGGAAAGTCCCCAGGCTCCCCAGCAGGCAGAAGTATGCAAAGCATGCATCTCAATTAGTCAGCAACCAGGTGTGGAAAGTCCCCAGGCTCCCCAGCAGGCAGAAGTATGCAAAGCATGCATCTCAATTAGTCAGCAACCATAGTCCCGCCCCTAACTCCGCCCATCCCGCCCCTAACTCCGCCCAGTTCCGCCCATTCTCCGCCCCATGGCTGACTAATTTTTTTTATTTATGCAGAGGCCGAGGCCGCCTCGGCCTCTGAGCTATTCCAGAAGTAGTGAGGAGGCTTTTTTGGAGGCCTAGGCTTTTGCAAA>sv40.fa
$ seqkit seq -s sv40.fa|awk '{for(i=1;;i++){delete a;for(j=1;j<=length-i+1;j++)a[substr($0,j,i)][j];found=0;for(x in a){if(length(a[x])>1){found=1;for(y in a[x])print i,y,y+length(x)-1,x}}if(!found)exit}}'|sort -srnk1,1|awk 'NR>1&&$1!=x{exit}{x=$1}1'
72 1 72 GGTGTGGAAAGTCCCCAGGCTCCCCAGCAGGCAGAAGTATGCAAAGCATGCATCTCAATTAGTCAGCAACCA
72 73 144 GGTGTGGAAAGTCCCCAGGCTCCCCAGCAGGCAGAAGTATGCAAAGCATGCATCTCAATTAGTCAGCAACCA
From the output above, you can see that the SV40 promoter sequence starts with a tandem repeat where the same 72-base segment is repeated twice in a row.
This finds palindromes, which are sequences that are identical to their reverse complement:
$ x=$(seqkit replace -p'.* ([^ ]*),.*' -r\$1 sars2.fa|seqkit replace -sip'a*$');paste <(seqkit fx2tab<<<"$x"|cut -f-2) <(seqkit seq -rp<<<"$x"|seqkit seq -s)|awk -F\\t '{max=length($2);for(len=startlen;;len+=2){found=0;for(i=1;i<=max-len+1;i++){fw=substr($2,i,len);rev=substr($3,max-len-i+2,len);if(fw==rev){found=1;print len,i,fw,$1}}if(!found)next}}' startlen=16
16 5747 ACTGGTAATTACCAGT Wuhan-Hu-1
16 6089 AACCAGTTAACTGGTT Wuhan-Hu-1
16 22901 GGTAATTATAATTACC Wuhan-Hu-1
18 5746 CACTGGTAATTACCAGTG Wuhan-Hu-1
20 5745 ACACTGGTAATTACCAGTGT Wuhan-Hu-1
This finds mirror repeats, which are sequences that are identical to their reversed form without taking a complement of the nucleotides:
$ seqkit replace -p'.* ([^ ]*),.*' -r\$1 sars2.fa|seqkit replace -sip'a*$'|seqkit fx2tab|awk -F\\t '{for(len=startlen;;len++){found=0;for(i=1;i<=length($2)-len+1;i++){fw=substr($2,i,len);rev="";for(j=len;j>=1;j--)rev=rev substr(fw,j,1);if(fw==rev){found=1;print len,i,fw,$1}}if(found==0&&prevfound==0)next;prevfound=found}}' startlen=16
16 9979 CAATGACTTCAGTAAC Wuhan-Hu-1
18 9978 TCAATGACTTCAGTAACT Wuhan-Hu-1
20 9977 CTCAATGACTTCAGTAACTC Wuhan-Hu-1
This finds all direct repeats of 15 bases:
$ seqkit head -n2 sarbe.fa|seqkit seq -g|seqkit replace -p'(,|surface|ORF|orf).*'|seqkit replace -sip'a*$'|seqkit fx2tab|awk -F\\t '{l=length($2);delete count;delete pos;for(i=1;i<=l-len+1;i++){motif=substr($2,i,len);count[motif]++;pos[motif][i]}for(i in count)if(count[i]>1){o=i;for(j in pos[i])o=o" "j;print NR,o";"$1}}' len=15|awk '$2!~/[nN]/'|sort -nk3|sort -snk1,1|cut -d\ -f2-|sed 's/ /;/'
cacttacgcagtata;76 9741;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
acttacgcagtataa;77 9742;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
aaaagaaaaagactg;1092 29142;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
aaagaaaaagactga;1093 29143;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
agtacggaagtttct;7889 23609;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
gtacggaagtttctg;7890 23610;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
taaacgaacatgaaa;27133 27639;OR233295.1 Horseshoe bat sarbecovirus isolate Rt17DN420
aaaagaaaaagactg;1108 29181;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
aaagaaaaagactga;1109 29182;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
gaagaagaagaagaa;3159 3162;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
aagaagaagaagaag;3160 3163;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
agaagaagaagaaga;3161 3164;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
ctaaacgaacatgaa;21430 27674;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
taaacgaacatgaaa;27169 27675;OR233321.1 Horseshoe bat sarbecovirus isolate Rt22QT53
This finds tandem repeats, which are direct repeats that occur twice in a row with no bases in between. The columns are length, starting position, repeated motif, and sequence name. Tandem repeats are rare in sarbecoviruses, so here I only found two tandem repeats longer than 10 bases in sarbecoviruses:
$ seqkit seq -g sarbe.fa|seqkit grep -sivp n|seqkit replace -p'(,|surface|ORF|orf).*'|seqkit replace -sip'a*$'|seqkit fx2tab|awk -F\\t '{l=length($2);for(len=10;;len++){found=0;delete count;delete pos;for(i=1;i<=l-len+1;i++){motif=substr($2,i,len);count[motif]++;pos[motif][i]}for(i in count)if(count[i]>1){found=1;o="";for(j in pos[i])if((j+len)in pos[i]&&i!~/^[aA]*$|[nN]/)print NR,len,j,(j+len),i";"$1}if(!found)next}}'
21 1901 tgcagcaaaccactcaattcc;OR233291.1 Horseshoe bat sarbecovirus isolate Ra21CB8
21 1902 gcagcaaaccactcaattcct;OR233291.1 Horseshoe bat sarbecovirus isolate Ra21CB8
21 1903 cagcaaaccactcaattcctg;OR233291.1 Horseshoe bat sarbecovirus isolate Ra21CB8
21 5 tccaggaaaagccaaccaacc;DQ640652.1 SARS coronavirus GDH-BJH01
21 6 ccaggaaaagccaaccaacct;DQ640652.1 SARS coronavirus GDH-BJH01
21 7 caggaaaagccaaccaacctc;DQ640652.1 SARS coronavirus GDH-BJH01
15 29288 aaaaaaaaaaaaaac;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29289 aaaaaaaaaaaaaca;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29290 aaaaaaaaaaaacaa;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29291 aaaaaaaaaaacaaa;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29292 aaaaaaaaaacaaaa;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29293 aaaaaaaaacaaaaa;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29294 aaaaaaaacaaaaaa;OQ401249.1 MAG: Sarbecovirus RfGB02
15 29295 aaaaaaacaaaaaaa;OQ401249.1 MAG: Sarbecovirus RfGB02
The code below shows how many mutations closer each GISAID sample is to RaTG13 than to Wuhan-Hu-1. There is one sample from Guangdong which has five mutations relative to Wuhan-Hu-1 but all of them are shared with RaTG13. The five mutations are the three mutations of proCoV2/WA1 in addition to two additional mutations, even though the collection date of the sample is in late February, so the two additional mutations might be reversions that occurred due to chance:
$ curl https://sars2.net/f/gisaid2020.tsv.xz|xz -dc>gisaid2020.tsv
$ grep RaTG13 gisaid2020.tsv|cut -f12|tr , \\n|awk -F\\t 'NR==FNR{a[$0];next}$11<10{split($12,b,",");n=0;for(i in b)n+=b[i]in a?1:-1;print n FS$0}' - gisaid2020.tsv|awk '$1>=3'|sort -rn|cut -f1,2,4-8,12-13|tr \\t \||head
5|EPI_ISL_413892|2020-03-09|2020-02-28|A|China|Guangdong|5|C8782T,C18060T,C21711T,C24382T,T28144C
3|EPI_ISL_855195|2021-01-19|2020-03-15|A|USA|New York|3|C8782T,C18060T,T28144C
3|EPI_ISL_671375|2020-12-02|2020-03-20|A|Ireland|Kerry|3|C8782T,C18060T,T28144C
3|EPI_ISL_654866|2020-11-25|2020-01-31|A|Vietnam|Ho Chi Minh City|3|C8782T,T28144C,C29095T
3|EPI_ISL_653255|2020-11-23|2020-03-04|A.1|USA|Florida|7|C6286T,C8782T,C17010T,C17747T,A17858G,C18060T,T28144C
3|EPI_ISL_653254|2020-11-23|2020-03-04|A.1|USA|Florida|7|C6286T,C8782T,C17010T,C17747T,A17858G,C18060T,T28144C
3|EPI_ISL_604189|2020-10-30|2020-03-14|A.1|USA|California|7|C6286T,C8782T,C17010T,C17747T,A17858G,C18060T,T28144C
3|EPI_ISL_596386|2020-10-23|2020-01-19|A|USA|Washington|3|C8782T,C18060T,T28144C
3|EPI_ISL_582763|2020-10-16|2020-03-27|A.5|Spain|Basque Country|3|C8683T,C8782T,T28144C
3|EPI_ISL_522942|2020-08-26|2020-08-06|A|Mexico|Mexico City|3|C8782T,C18060T,T28144C
brew install seqkit mafft
wget https:/sars2.net/f/pid.cpp;g++ pid.cpp -O3 -o pid
# percentage identity matrix of 529 sarbecovirus sequences
wget https://sars2.net/f/sarbe.pid
# matrix sort grep (for a matrix with row names and column names, print the values of a column whose name matches a substring and sort by the column)
masog()(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~x)break;next}{print$i,$1}' x="$1" "${2--}"|sort -n)
# efetch multi (download list of accessions from STDIN)
emu()(curl -sd "id=$(paste -sd, -)" "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1:-nuccore}&rettype=${2:-fasta}&retmode=$3")
# align spike proteins of Tor2 and sequences with 95-98% identity to Tor2
masog Tor2, sarbe.pid|awk '$1==100||($1<=98&&$1>=95){print$2}'|emu '' fasta_cds_na|seqkit grep -nrp 'gene=S|spike|surface'|sed 's/_cds_.*//;s/lcl|//'|seqkit fx2tab|awk 'NR==FNR{a[$1]=$0;next}{print">"a[$1]"\n"$2}' <(cut -f1 sarbe.pid|cut -d, -f1) -|mafft --thread 4 --quiet ->temp.fa;./pid<temp.fa>temp.pid
# install.packages("BiocManager");BiocManager::install("Biostrings")
library(ggplot2)
library(colorspace)
library(Biostrings)
t=readDNAStringSet("temp.fa")
names(t)=sub("^.*? ","",sub(",.*","",names(t)))
targ=grep("Tor2",names(t),value=T)
step=300
max=width(t)[1]
ranges=lapply(seq(1,max,step),\(i)c(i,min(i+step-1,max)))
heat=100*sapply(ranges,\(x)as.matrix(stringDist(subseq(t,x[1],x[2]),"hamming"))[targ,]/(x[2]-x[1]))
# heat=100*sapply(ranges,\(x){block=t[,x[1]:x[2]];rowSums(block[targ,]!="-"&block!="-"&block[targ,]==block)/ncol(block)}) # don't count positions where either sequence has a gap
# colnames(heat)=sapply(ranges,paste,collapse="-") # display nucleotide positions instead of amino acid positions
colnames(heat)=apply(ranges,1,\(x)paste0((x[1]-1)/3+1,"-",x[2]/3))
dist=100-read.table("temp.pid",sep="\t",row.names=1,header=T)
pick=!grepl("Mutant|related coronavirus isolate",rownames(heat))
heat=heat[pick,]
dist=dist[pick,pick]
hc=as.hclust(reorder(as.dendrogram(hclust(as.dist(dist))),cmdscale(dist,1)))
rownames(heat)=sub("Severe acute respiratory syndrome","SARS",rownames(heat))
rownames(heat)=sub(" (ORF|surface).*","",rownames(heat))
rownames(heat)=sub(" isolate "," ",rownames(heat))
pheatmap::pheatmap(
heat,
filename="0.png",
clustering_callback=\(...)hc,
cluster_cols=F,
legend=F,
cellwidth=18,
cellheight=14,
fontsize=9,
treeheight_row=80,
treeheight_column=80,
border_color=NA,
display_numbers=T,
number_format="%.0f",
fontsize_number=8,
number_color=matrix(ifelse(heat>.8*max(heat),"white","black"),nrow(heat))[hc$order,],
na_col="white",
breaks=seq(0,max(heat),,256),
colorRampPalette(hex(HSV(c(210,210,210,160,110,60,40,20,0,0,0),c(0,.25,rep(.5,9)),c(rep(1,9),.5,0))))(256)
)
# system(paste0("convert -gravity north \\( -splice 0x26 -pointsize 46 label:'Target: ",targ," (percentage of nucleotide changes in each ",step," nt block)' \\) 0.png -append 1.png"))
system(paste0("convert -gravity north \\( -splice 0x26 -pointsize 46 label:'Target: Tor2 (percentage of nucleotide changes in each ",(step/3)," aa block of spike protein)' \\) 0.png -append 1.png"))
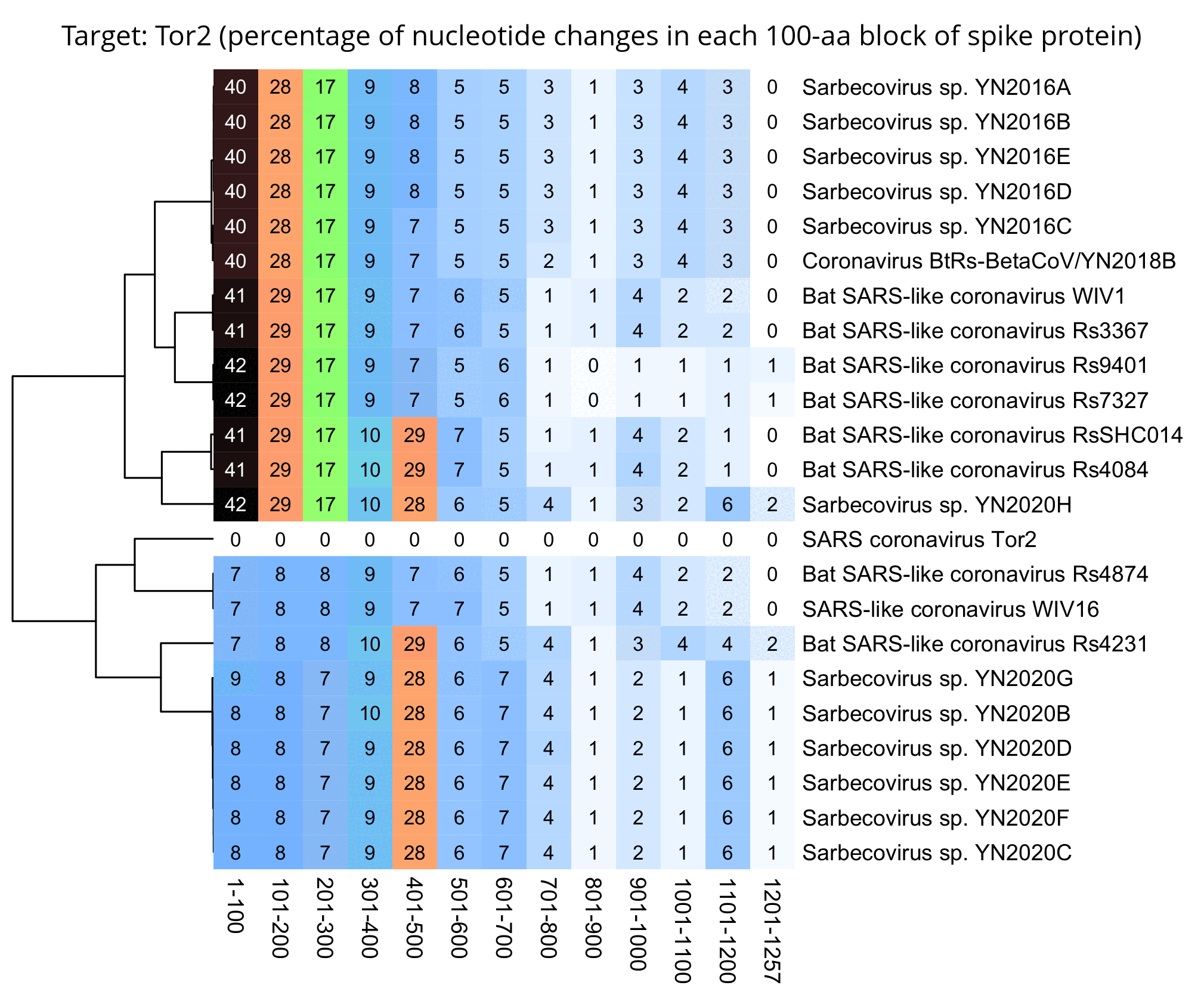
The heatmap above shows that you can divide the viruses that are the closest to SARS1 to four groups based on the N-terminal domain and receptor binding motif. WIV1 has a different NTD but similar RBM, Rs4231 has a different RBM but similar NTD, in SHC014 both NTD and RBM are different, and in WIV16 both the NTD and RBM are similar.
This image from a patent by Ralph Baric et al. shows that the NTD is located approximately at positions 1-318 and the RBM is located at approximately positions 438-509: [https://patents.google.com/patent/US9884895B2/en]
Valentin Bruttel has said that because the RdRp of RaTG13 has only 4 amino acid changes from Wuhan-Hu-1 so that there's about 99.5% amino acid identity, the rest of the genome of RaTG13 may have been artificially manipulated to have a higher distance to SARS2, because the whole genome of RaTG13 has only about 96.2% identity with SARS2 on the nucleotide level. [https://x.com/VBruttel/status/1722617170663010438, https://www.youtube.com/watch?v=EuuY94tsbls&t=1h1m58s]
However RdRp is a conserved region, so the number of amino acid changes from Wuhan-Hu-1 in the RdRp is only 1 in all five BANAL viruses and RpYN06, and it's 3 in some of the new Vietnamese sequences from Hassanin et al. 2023, and it's only 34 even in Tor2. So since the RdRp of Wuhan-Hu-1 is 932 aa long, the RdRp of Tor2 has about 96.4% amino acid identity to Wuhan-Hu-1. (The RdRp might have slightly different starting and ending positions in different sarbecoviruses, but I'm using the positions corresponding to Wuhan-Hu-1 in my calculation.)
Here the first column shows the number of amino acid changes from Wuhan-Hu-1 in the RdRp, the second column shows the number of nucleotide changes in the RdRp, and the third column shows the nucleotide percentage identity with Wuhan-Hu-1 in the whole genome:
$ wget -q https://sars2.net/f/sarbe.{pid,fa.xz}
$ range=$(seqkit grep -nrp Wuhan-Hu-1 sarbe.fa.xz|seqkit seq -s|gawk -F '' '{for(i=1;i<=length;i++){if($i=="-")gaps++;if(i-gaps==13442)printf"%s:",i;if(i-gaps==16237)print i}}')
$ awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~/Hu-1/)break;next}$1!=x{print$i,$1}' sarbe.pid|awk '($1>=90&&$1<100)||/Tor2/{print$2}'|seqkit grep -f- sarbe.fa.xz|cat <(seqkit grep -nrp Hu-1 sarbe.fa.xz) -|seqkit subseq -r$range|mafft --thread 4 ->temp.fa
[...]
$ x=$(seqkit seq -n temp.fa|sed 's/,.*//;s/ \(ORF\|surface\).*//;s/ isolate / /'|paste - <(seqkit seq -s temp.fa|sed 's/./&&/26')|seqkit tab2fx)
$ dist1()(seqkit fx2tab "$@"|awk -F\\t 'NR==1{split($2,a,"");l=length(a);next}{split($2,b,"");d=0;for(i=1;i<=l;i++)if(a[i]!="-"&&b[i]!=""&&a[i]!=b[i])d++;print d,$1}')
$ dist1<<<"$x"|sed $'s/ /\t/;s/ /\t/'|paste <(seqkit translate<<<"$x"|dist1|cut -d\ -f1) -|sed 's/,.*//;s/ \(ORF\|surface\).*//;s/ isolate / /'|awk 'NR==FNR{a[$2]=$1;next}{$3=sprintf("%.1f",a[$3])}1' {,O}FS=\\t <(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~/Hu-1/)break;next}{sub(/ .*/,"",$1);print$i FS$1}' sarbe.pid) -|sort -nk2|column -ts$'\t'
1 43 96.0 Bat coronavirus BANAL-20-236/Laos/2020
1 44 97.0 Bat coronavirus BANAL-20-52/Laos/2020
1 46 94.8 Betacoronavirus sp. RpYN06 strain bat/Yunnan/RpYN06/2020
1 48 93.9 Bat coronavirus BANAL-20-116/Laos/2020
1 48 93.9 Bat coronavirus BANAL-20-247/Laos/2020
1 48 96.1 Bat coronavirus BANAL-20-103/Laos/2020
4 61 96.2 Bat coronavirus RaTG13
7 85 92.8 Horseshoe bat sarbecovirus Ra22QT106
7 85 92.8 Horseshoe bat sarbecovirus Ra22QT137
7 85 92.8 Horseshoe bat sarbecovirus Ra22QT77
8 86 92.8 Horseshoe bat sarbecovirus Ra22QT135
3 94 90.6 Bat coronavirus RacCS253
3 94 90.9 Bat coronavirus RacCS271
3 94 91.9 Bat coronavirus RacCS203
5 241 90.4 MAG: Pangolin coronavirus GD/P79-9/2019
5 242 90.3 Pangolin coronavirus MP789
34 320 79.7 SARS coronavirus Tor2
34 329 92.3 Bat SARS-like virus BtSY2
34 332 91.0 Sarbecovirus sp. PrC31
34 333 92.2 Horseshoe bat sarbecovirus Rp22DB159
37 335 90.1 Sarbecovirus sp. YN2021
The RdRp of SARS2 is very distant from BtSY2, PrC31, and YN2021, but that's because they are recombinant viruses where some regions are similar to SARS2-like viruses and other regions are similar to SARS1-like viruses, but the RdRp is one of the regions that is more similar to SARS1.
ORF1ab has a ribosomal slippage where the frame is shifted one base backwards at the start of ORF1b, so that the last base of ORF1a gets duplicated as the first base of ORF1b. In Wuhan-Hu-1 the first 26 bases of the RdRp come from ORF1a and the rest comes from ORF1b, so the code above inserts the last base of ORF1a to position 27, because otherwise the amino acid translation of the RdRp gets messed up:
$ seqkit subseq -r 13442:16237 sars2.fa|seqkit translate|seqkit subseq -r1:60 # the translation is messed up because of ribosomal slippage >MN908947.3 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome SADAQSFLNGFAV*VQPVLHRAAQALVLMSYTGLLTSTMIK*LVLLNS*KLIVVASKKRT $ seqkit seq -s sars2.fa|cut -c 13442-16237|(echo \>a;sed 's/./&&/26')|seqkit translate|seqkit subseq -r1:60 # insert last base of ORF1a to start of ORF1b >a SADAQSFLKRVCGVSAARLTPCGTGTSTDVVYRAFDIYNDKVAGFAKFLKTNCCRFQEKD $ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&rettype=fasta&id=YP_009725307'|seqkit subseq -r1:60 # compare to RdRp sequence from GenBank >YP_009725307.1 RNA-dependent RNA polymerase [Severe acute respiratory syndrome coronavirus 2] SADAQSFLNRVCGVSAARLTPCGTGTSTDVVYRAFDIYNDKVAGFAKFLKTNCCRFQEKD
When Kevin McKernan sequenced Pfizer vaccine vials, he found that the reverse strand of the spike protein sequence had a 1,252-aa open reading frame. [https://anandamide.substack.com/p/what-do-you-make-of-this-sars1-patent] In an RNA sequence it wouldn't matter but the vaccines were contaminated with double-stranded DNA. McKernan has been calling the long ORF on the reverse strand the "spidroin" because its closest hit on UniProt was a spidroin protein which is the main protein in spider silk, even though the similarity was not very high and the match was almost certainly accidental.
If you for search for ORFs in all three frames of the reverse strand of Wuhan-Hu-1, the longest ORF is only 150 amino acids long:
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947.3'>sars2.fa
$ r=$(seqkit seq -rp sars2.fa|seqkit seq -s);for x in $r ${r:1} ${r:2};do sed 's/.../& /g'<<<$x|grep -Po '(ATG|CTG|TTG).*?(?=(TAA|TAG|TGA))'|tr -d \ ;done|awk '{print">"NR}1'|seqkit translate|seqkit seq -gm4|seqkit stat
file format type num_seqs sum_len min_len avg_len max_len
- FASTA Protein 669 13,036 4 19.5 150
However the spike sequence in the Pfizer jabs was codon-optimized, which eliminated some of the stop codons on the reverse strand.
This GitHub repository includes a full sequence for a "spike-encoding contig assembled from BioNTech/Pfizer BNT-162b2 vaccine": https://github.com/NAalytics/Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273/. When I searched for ORFs on all 3 frames on the reverse strand, my longest ORF was 1,341 aa:
$ curl https://github.com/NAalytics/Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273/raw/refs/heads/main/Figure1Figure2_032321.fasta|seqkit grep -nrp Pfizer>pfizerspike.fa
$ r=$(seqkit seq -rp pfizerspike.fa|seqkit seq -s);for x in $r ${r:1} ${r:2};do sed 's/.../& /g'<<<$x|grep -Po '(ATG|CTG|TTG).*?(?=(TAA|TAG|TGA))'|tr -d \ ;done|awk '{print">"NR}1'|seqkit translate|seqkit sort -lr|seqkit head -n1
>57
LLRAWEVSGTSRGGEWGRWRWEHTWDPRSGETRGTQDGKGAASIACMQYQLESSCVVQFH
ALQHGLRIVLVELAAAATATTALQAATAAGHATQHDCDHHDGNQSGDKAQPDVPGPLDVL
LVLPQFLQVDQALVQILGHLVQPVDLFLDVHDAGIDSADIAQVHVGACVVLKVLVQFLFE
AVQLGLQRVVHGIVHNADHDVAVARHEGVVGGDDLGLVEVPLCHEPMGAVGHEHAFSRKV
GFAVVADGWSGGEILLLSGHICHVQKHHAVRGRLREAHQVVALAAKVHSLALAQHTLRHL
GGGQIGRGSNLGGSDQLLGHVCLEALQSACDQSVDLHLGLRRVQSAQDIVQHRADGAEVG
GQLLDQGVQCLGILVDHVLQLSQGACCAAQAVLDLADGAVELVGDQLLVLVQHILGHSDA
VEPVGHLHSKGDLQSGACSKCPAACDCAGQQGRCVLGDHLIGQQRRQHCQSVKLLGANQI
PGGNVAQTIAILLDEAGVGQCHFVEQQVLDEAPLAGLARIGQNLAEIEAAEVLDRRGLVD
LLHLGEHLLGVLVLFHGDPCQGSIQLGAEAAVLQQQVGALGGIAADVHGAVHAGLGHGHR
QDLCGHADGEVGGDSDRVVGVGHAVLGAQRHCVGNDALAGHASGSPRAVCLCLVAGADSS
ADGDVALVAIVHVLGSDQTAGSGLKHIAAGGVHPPCRCQLIGVNGHGHFGTVHVLVQHCH
LIAGVGARGDHRHSAEAARGDVQDFQCLGISNGVCGIGDIPAKLLEWQELLVALCQHAGA
GQAVEVEVHAFVLHEIGAFLRAAHCGRGMQQFEAQHHHSVGLIAHAICGLKAVGLQWEVA
VEAFHAVTRGAAGLIDLGGDVPLEGLQIGLPEQPVQVIVVAADFGVQVVAVPGNHTAGEV
VGQLVVVVGDLACLSRGNLPHFISPDHEAVGVHVCEAQVVQLGRGHAVALEGAEAGGVVQ
HGVVGHAIADPLPVPGVHRGESGGIEHLAEGAQIGDIGEPHDGFGGLHPEVAGLVDALFH
GEGLQGALCLAQRIQSTIHGVGDGAVLVVLQQEGSRLQVAHIVSGGTSCPSAAAIARCQV
ASVQGQQCLKPGDVDADGQIHQGFQSREALRQIPHEVDRGVLAVDLEVAVDVLKHELAQV
LEVALLAFQVHQERLGHVLEGAVVGAAVHPELAFHPALVVLVVVDAQEGVVAELELAHFD
DHVGGVVHDQQALGLAVQCGAEDPASDDVGLLGAGKVHPVVEGQHGVVESLGAIGAGHVD
GVEPGHVAEERQEQVLGRVQHAGSEHLVGVVHASGKAVGVGWRQLCSGGQVHTLAGHQRQ
QHQEHEHGGGFSLSLWGPEEY
I tried doing a BLAST search for the reverse ORF at UniProt: https://www.uniprot.org/blast. My best two matches were a protein from algae and a protein from a cockroach, but the spider silk protein found by McKernan was only the third-best match. (The match to the spider silk protein is of course accidental, but for example Ana Maria Mihalcea is now saying that kalamari clots contain spidroin, vaccines have nanobots that contain spidroin, and chemtrails contain spidroin.)
NCBI's Sequence Read Archive has a little-known "SRA Database Backend" API: https://trace.ncbi.nlm.nih.gov/Traces/sra-db-be/. It can be used to retrieve the full STAT results of an SRA run, which are not displayed anywhere on the SRA's website.
This displays the most abundant leaf nodes in the metagenomic sequencing run of Wuhan-Hu-1:
$ curl -Ls "https://trace.ncbi.nlm.nih.gov/Traces/sra-db-be/run_taxonomy?acc=SRR10971381&cluster_name=public">SRR10971381.stat $ jq -r '[.[]|.tax_table[]|.parent]as$par|[.[]|.tax_table[]|select(.tax_id as$x|$par|index($x)|not)]|sort_by(-.total_count)[]|((.total_count|tostring)+";"+.org)' SRR10971381.stat|head 171042;Prevotella salivae F0493 155602;Prevotella veroralis F0319 89078;Prevotella scopos JCM 17725 57886;Prevotella melaninogenica D18 54238;Severe acute respiratory syndrome coronavirus 2 51814;Leptotrichia sp. oral taxon 212 41272;Prevotella nanceiensis DSM 19126 = JCM 15639 33716;Homo sapiens 28813;Prevotella veroralis DSM 19559 = JCM 6290 23601;Leptotrichia hongkongensis
This displays viral leaf nodes along with the chain of ancestors of each node:
$ jq -r '.[]|.tax_table[]|[.org,.total_count,.tax_id,.parent]|@tsv' SRR10971381.stat|awk -F\\t '{out[$3]=$0;parent[$3]=$4;name[$3]=$1;nonleaf[$4]}END{for(i in out){j=parent[i];chain="";do{chain=(chain?chain":":"")name[j];j=parent[j]}while(j!="");print out[i]"\t"!(i in nonleaf)"\t"chain}}'|sort -nr|grep Virus|sort -t$'\t' -rnk2|awk -F\\t '$5{print$2 FS$1" - "$6}'|head -n20|column -ts$'\t'
54238 Severe acute respiratory syndrome coronavirus 2 - Severe acute respiratory syndrome-related coronavirus:Sarbecovirus:Betacoronavirus:Orthocoronavirinae:Coronaviridae:Cornidovirineae:Nidovirales:Riboviria:Viruses
693 CRESS virus sp. - CRESS viruses:unclassified ssDNA viruses:unclassified DNA viruses:unclassified viruses:Viruses
159 uncultured virus - environmental samples:Viruses
20 Parvovirus NIH-CQV - unclassified Parvovirinae:Parvovirinae:Parvoviridae:Viruses
15 Curvibacter phage P26059B - unclassified Autographivirinae:Autographivirinae:Podoviridae:Caudovirales:Viruses
13 Pseudomonas phage HU1 - unclassified Podoviridae:Podoviridae:Caudovirales:Viruses
12 Bat coronavirus RaTG13 - Severe acute respiratory syndrome-related coronavirus:Sarbecovirus:Betacoronavirus:Orthocoronavirinae:Coronaviridae:Cornidovirineae:Nidovirales:Riboviria:Viruses
9 Iodobacter phage PhiPLPE - Iodobacter virus PLPE:Iodovirus:Myoviridae:Caudovirales:Viruses
9 Bat SARS-like coronavirus - Severe acute respiratory syndrome-related coronavirus:Sarbecovirus:Betacoronavirus:Orthocoronavirinae:Coronaviridae:Cornidovirineae:Nidovirales:Riboviria:Viruses
8 Rhodoferax phage P26218 - unclassified Podoviridae:Podoviridae:Caudovirales:Viruses
8 Prokaryotic dsDNA virus sp. - unclassified dsDNA viruses:unclassified DNA viruses:unclassified viruses:Viruses
8 Pangolin coronavirus - unclassified Betacoronavirus:Betacoronavirus:Orthocoronavirinae:Coronaviridae:Cornidovirineae:Nidovirales:Riboviria:Viruses
5 uncultured cyanophage - environmental samples:unclassified bacterial viruses:unclassified viruses:Viruses
5 Rimavirus - Siphoviridae:Caudovirales:Viruses
4 Genomoviridae sp. - unclassified Genomoviridae:Genomoviridae:Viruses
3 Streptococcus phage Javan363 - unclassified Siphoviridae:Siphoviridae:Caudovirales:Viruses
3 Sewage-associated circular DNA virus-14 - unclassified viruses:Viruses
3 RDHV-like virus SF1 - unclassified viruses:Viruses
3 Paramecium bursaria Chlorella virus NW665.2 - unclassified Chlorovirus:Chlorovirus:Phycodnaviridae:Viruses
3 Microviridae sp. - unclassified Microviridae:Microviridae:Viruses
I tried looking at the STAT results of 1000 random SRA runs which matched the phrase "wastewater metagenome 2023[dp]". These were the most common leaf nodes in the STAT hits. SARS2 has by far the highest number of reads but it's probably because in many runs they were specifically targeting SARS2:
$ curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh|sh
[...]
$ esearch -db sra -query 'wastewater 2023[dp]'|efetch -format runinfo>waste.runinfo
$ mkdir waste;sed 1d waste.runinfo|gshuf -n1000|cut -d, -f1|parallel -j10 -q curl -m 30 -s 'https://trace.ncbi.nlm.nih.gov/Traces/sra-db-be/run_taxonomy?acc={}&cluster_name=public' -o waste/{}.stat
$ for f in waste/*;do jq -r '.[]|.tax_table[]|[.org,.total_count,.tax_id,.parent]|@tsv' $f|awk -F\\t '{out[$3]=$0;parent[$3]=$4;name[$3]=$1;nonleaf[$4]}END{for(i in out){j=parent[i];chain="";do{chain=(chain?chain":":"")name[j];j=parent[j]}while(j!="");print out[i]"\t"!(i in nonleaf)"\t"chain}}'|awk -F\\t '$5{print$2 FS$1" - "$6}';done>temp
$ grep Viruses temp|awk -F\\t '{a[$2]+=$1}END{for(i in a)print a[i]"\t"i}'|sort -rn|head -n20|column -ts$'\t'
139686660 Severe acute respiratory syndrome coronavirus 2 - Severe acute respiratory syndrome-related coronavirus:Sarbecovirus:Betacoronavirus:Orthocoronavirinae:Coronaviridae:Cornidovirineae:Nidovirales:Pisoniviricetes:Pisuviricota:Orthornavirae:Riboviria:Viruses
321282 Caudoviricetes sp. - unclassified Caudoviricetes:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
131071 Sapovirus Hu/Chiba/010451S/2001 - Sapovirus isolates:Sapporo virus:Sapovirus:Caliciviridae:Picornavirales:Pisoniviricetes:Pisuviricota:Orthornavirae:Riboviria:Viruses
100819 EBPR siphovirus 1 - unclassified Caudoviricetes:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
85839 Sapovirus GI.1 - Sapovirus GI:Sapporo virus:Sapovirus:Caliciviridae:Picornavirales:Pisoniviricetes:Pisuviricota:Orthornavirae:Riboviria:Viruses
54246 Bacteriophage sp. - unclassified bacterial viruses:Viruses
22293 Microviridae sp. - unclassified Microviridae:Microviridae:Petitvirales:Malgrandaviricetes:Phixviricota:Sangervirae:Monodnaviria:Viruses
20054 Sapovirus GI.2 - Sapovirus GI:Sapporo virus:Sapovirus:Caliciviridae:Picornavirales:Pisoniviricetes:Pisuviricota:Orthornavirae:Riboviria:Viruses
12342 Vibrio phage VvAW1 - unclassified Caudoviricetes:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
9327 Gordonia phage GMA7 - Getseptimavirus GMA7:Getseptimavirus:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
8646 Sapovirus Hu/S07026/2006/DNK - Sapovirus isolates:Sapporo virus:Sapovirus:Caliciviridae:Picornavirales:Pisoniviricetes:Pisuviricota:Orthornavirae:Riboviria:Viruses
7626 Mycobacterium phage Myrna - Myrnavirus myrna:Myrnavirus:Ceeclamvirinae:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
5031 Microvirus sp. - unclassified Microviridae:Microviridae:Petitvirales:Malgrandaviricetes:Phixviricota:Sangervirae:Monodnaviria:Viruses
4684 Caulobacter phage CcrRogue - Poindextervirus rogue:Poindextervirus:Dolichocephalovirinae:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
4269 Tombunodavirus UC1 - unclassified viruses:Viruses
4185 Aeromonas phage phiA005 - unclassified Caudoviricetes:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
3827 Sphaerotilus phage vB_SnaP-R1 - unclassified Krylovirinae:Krylovirinae:Autographiviridae:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
2891 uncultured human fecal virus - environmental samples:Viruses
2762 Brevundimonas phage vB_BsubS-Delta - unclassified Caudoviricetes:Caudoviricetes:Uroviricota:Heunggongvirae:Duplodnaviria:Viruses
2722 Murine coronavirus - Embecovirus:Betacoronavirus:Orthocoronavirinae:Coronaviridae:Cornidovirineae:Nidovirales:Pisoniviricetes:Pisuviricota:Orthornavirae:Riboviria:Viruses
This downloads metadata for a global subsample of about 3000 SARS2 sequences from NextStrain, and it selects nucleotide changes which appear in over half of all sequences where the variant is listed as Alpha:
$ curl https://data.nextstrain.org/files/ncov/open/global/metadata.tsv.xz|xz -dc>global.tsv
$ awk -F\\t '$34=="Alpha"' global.tsv|cut -f56|awk '{split($0,z,",");for(i in z)a[z[i]]++}END{for(i in a)if(a[i]>NR/2)print substr(i,2),i}'|sort -n|cut -d\ -f2
C241T
C913T
C3037T
C3267T
C5388A
C5986T
T6954C
C14408T
C14676T
C15279T
T16176C
A23063T
C23271A
A23403G
C23604A
C23709T
T24506G
G24914C
C27972T
G28048T
A28111G
G28280C
A28281T
T28282A
G28881A
G28882A
G28883C
C28977T
I used only ATG as a start codon here but I'm not sure if I should've also included CTG or TTG as start codons, because all CDS sequences of SARS2 start with ATG and RNA viruses generally use ATG as a start codon.
This also includes overlapping ORFs where the ending position of a previous ORF is after the starting position of the next ORF.
$ curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947'>sars2.fa
$ seqkit locate -Prp 'ATG(...)*?(TAA|TAG|TGA)' sars2.fa|cut -f5-7|awk '{if(length($3)>50)$3=substr($3,1,50)"..."}1'|awk '{$2=$2" "(NR==1?"length":$2-$1+1)}1'|column -t
start end length matched
107 136 30 ATGCTTAGTGCACTCACGCAGTATAATTAA
266 13483 13218 ATGGAGAGCCTTGTCCCTGGTTTCAACGAGAAAACACACGTCCAACTCAG...
13526 13567 42 ATGTCGTATACAGGGCTTTTGACATCTACAATGATAAAGTAG
13625 13636 12 ATGACAATTTAA
13685 13759 75 ATGAAGAAACAATTTATAATTTACTTAAGGATTGTCCAGCTGTTGCTAAA...
13768 21555 7788 ATGGTACCACATATATCACGTCAACGTCTTACTAAATACACAATGGCAGA...
21536 25384 3849 ATGTTCTTGTTAACAACTAAACGAACAATGTTTGTTTTTCTTGTTTTATT...
25332 25448 117 ATGAAGACGACTCTGAGCCAGTGCTCAAAGGAGTCAAATTACATTACACA...
25393 26220 828 ATGGATTTGTTTATGAGAATCTTCACAATTGGAACTGTAACTTTGAAGCA...
26183 26281 99 ATGATGAACCGACGACGACTACTAGCGTGCCTTTGTAAGCACAAGCTGAT...
26245 26472 228 ATGTACTCATTCGTTTCGGAAGAGACAGGTACGTTAATAGTTAATAGCGT...
26523 27191 669 ATGGCAGATTCCAACGGTACTATTACCGTTGAAGAGCTTAAAAAGCTCCT...
27202 27387 186 ATGTTTCATCTCGTTGACTTTCAGGTTACTATAGCAGAGATATTACTAAT...
27359 27397 39 ATGAAGAGCAACCAATGGAGATTGATTAAACGAACATGA
27394 27759 366 ATGAAAATTATTCTTTTCTTGGCACTGATAACACTCGCTACTTGTGAGCT...
27756 27887 132 ATGATTGAACTTTCATTAATTGACTTCTATTTGTGCTTTTTAGCCTTTCT...
27868 27897 30 ATGAAACTTGTCACGCCTAAACGAACATGA
27894 28259 366 ATGAAATTTCTTGTTTTCTTAGGAATCATCACAACTGTAGCTGCATTTCA...
28274 29533 1260 ATGTCTGATAATGGACCCCAAAATCAGCGAAATGCACCCCGCATTACGTT...
29538 29570 33 ATGCAGACCACACAAGGCAGATGGGCTATATAA
29558 29674 117 ATGGGCTATATAAACGTTTTCGCTTTTCCGTTTACGATATATAGTCTACT...
29771 29806 36 ATGCTAGGGAGAGCTGCCTATATGGAAGAGCCCTAA
29806 29844 39 ATGTGTAAAATTAATTTTAGTAGTGCTATCCCCATGTGA
# efetch multi (download accessions from STDIN from GenBank)
emu()(curl -sd "id=$(paste -sd,)" "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1:-nuccore}&rettype=${2:-fasta}&retmode=$3")
# download envelope proteins of viruses with less than 10% distance to Wuhan-Hu-1 in the whole genome
awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~/Wuhan-Hu-1/)break;next}$i>90{print$1}' sarbe.pid|cut -d\ -f1|emu '' fasta_cds_aa|seqkit grep -nrp 'gene=E|protein=small envelo|protein=envelope'>env.aa
# awk consensus
acons()(gawk -F '' '{for(i=1;i<=NF;i++)a[i][$i]++}END{for(i in a){max=0;for(j in a[i])if(a[i][j]>max){max=a[i][j];o=j}printf"%s",o};print""}')
# highlight positions that are different from the first row
cohl()(awk 'NR==1{split($0,a,"");next}{split($0,b,"");for(i=1;i<=length;i++)printf"%s",b[i]==a[i]?b[i]:"\033[31m"b[i]"\033[0m";print""}' "$@")
# highlight positions that are different from the consensus using input with one sequence per line
cohl2()(x=$(cat);acons<<<"$x"|cohl - <(printf %s\\n "$x"))
# highlight positions that are different from the consensus using FASTA input
cohl3()(x=$(seqkit fx2tab);paste -d\ <(cut -f1 <<<"$x") <(cut -f2<<<"$x"|cohl2))
seqkit fx2tab env.aa|sed $'s/lcl|//;s/_prot_[^\t]*//'|awk -F\\t 'NR==FNR{a[$1]=$2;next}{print">"a[$1]"\n"$2}' <(seqkit seq -n sarbe.fa|sed 's/,.*//;s/ \(ORF\|surface\).*//;s/ .* /\t/') -|mafft --quiet -|cohl3|column -t

# awk consensus
acons()(gawk -F '' '{for(i=1;i<=NF;i++)a[i][$i]++}END{for(i in a){max=0;for(j in a[i])if(a[i][j]>max){max=a[i][j];o=j}printf"%s",o};print""}')
# highlight positions that are different from the first row
cohl()(awk 'NR==1{split($0,a,"");next}{split($0,b,"");for(i=1;i<=length;i++)printf"%s",b[i]==a[i]?b[i]:"\033[31m"b[i]"\033[0m";print""}' "$@")
# highlight positions that are different from the consensus using input with one sequence per line
cohl2()(x=$(cat);acons<<<"$x"|cohl - <(printf %s\\n "$x"))
brew install seqkit bowtie2
curl -sL sars2.net/f/sarbe.fa.xz|gzip -dc>sarbe.fa;seqkit seq -g sarbe.fa>sarbe.ungap
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta_cds_na&id=NC_045512'>sars2.na
bow()([[ -e $1.1.bt2 ]]||bowtie2-build --threads 3 "$1" "$1";bowtie2 --no-unal -p3 -x "$@")
seqkit grep -nrp =S sars2.na|seqkit subseq -r $[438*3-2]:$[508*3]|bow sarbe.ungap -fU- -a --score-min L,-1,-1|grep -v ^@|ruby -ane'puts [$F[2],$F[3],$F[3].to_i+$F[5].scan(/\d+(?=[MD])/).map(&:to_i).sum-1]*" "'|while read l m n;do seqkit grep -p $l sarbe.ungap|seqkit subseq -r $m:$n;done|seqkit grep -nrvp 'MP789 genomic sequence'|sed -E $'s/(,| surface| ORF1| genomic sequence).*//;s/.* />/'>rbm.fa
seqkit translate rbm.fa|seqkit seq -s|sed 's/./ & /g'|cohl2|sed s/31m/35m/g|paste -d\\n - <(seqkit seq -s rbm.fa|cohl2)|paste -d\ - <(seqkit seq -n rbm.fa|sed 'x;p;x')

This highlights positions that differ from the first sequence:
seqdif()(seqkit fx2tab|gawk -F\\t '{name[NR]=$1;split($2,z,"");for(i in z)seq[NR][i]=z[i];if(length($1)>max1)max1=length($1);if(length($2)>max2)max2=length($2)}END{for(i=1;i<=max2;i+=width){printf("%"(max1+1)"s","");for(pos=i;pos<i+width&&pos<=max2;pos+=10)printf(pos-i>width-10?"%s":"%-10s",pos);print "";for(j in seq){out=sprintf("%"max1"s ",name[j]);for(k=i;k<=max2&&k<i+width;k++)out=out (seq[j][k]==seq[1][k]?seq[j][k]:"\033[31m"seq[j][k]"\033[0m");print out}}}' "width=${1-60}")
emu()(curl -sd "id=$(paste -sd,)" "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1:-nuccore}&rettype=${2:-fasta}&retmode=$3")
printf %s\\n MN908947.{3..1} MN996532.{2,1}|emu|seqkit replace -p' .* ([^ ]*),.*' -r' $1'|seqkit subseq -r 1:200|mafft --quiet -|seqkit subseq -r 1:120 >temp
seqdif<temp
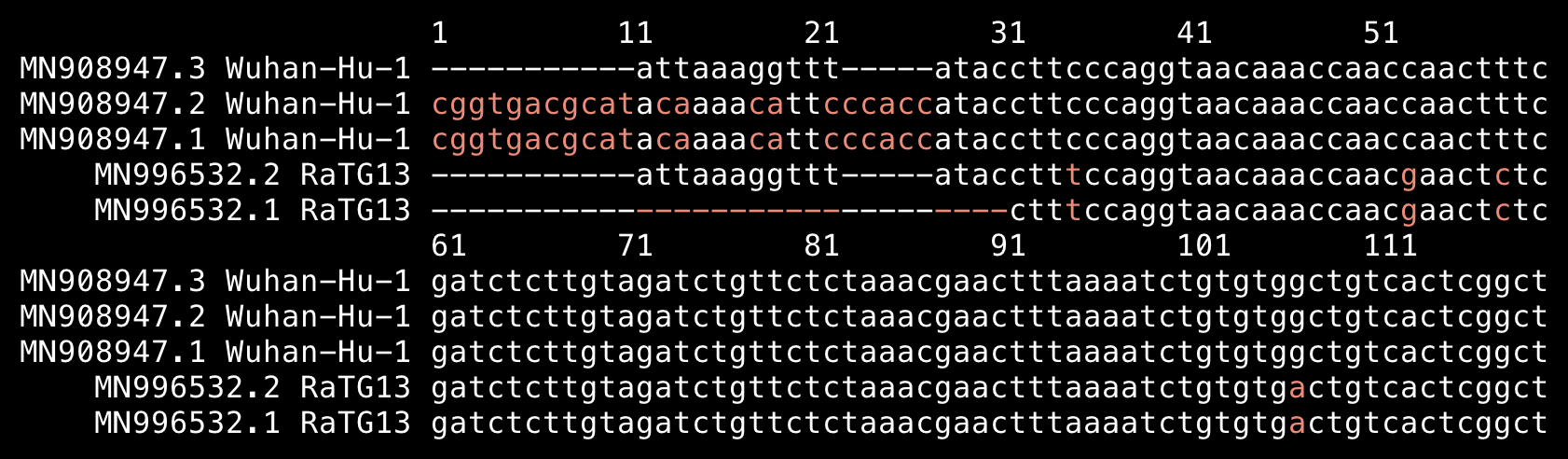
This uses a different color for each amino acid or nucleotide:
# # generate color palette
# Rscript --no-init-file -e 'hue=c(0,30,60,90,135,170,210,240,270,300)/360;x=colorspace::hex2RGB(c(hsv(hue,.45,.95),hsv(hue,.3,.85),hsv(hue,.45,.5)));write.table(255*attr(x,"coords"),col.names=F,row.names=F)'
aacolor()(printf %s\\n A:242:121:121 C:242:182:121 D:242:242:121 G:182:242:121 F:121:242:151 E:121:242:222 T:121:182:242 I:121:121:242 K:182:121:242 L:242:121:242 M:255:191:191 N:255:223:191 P:255:255:191 Q:223:255:191 R:191:255:207 S:191:255:244 H:191:223:255 V:191:191:255 W:223:191:255 Y:255:191:255 -:140:140:140|tr : \ )
seqcol()(seqkit seq -u|seqkit fx2tab|gawk -F\\t 'NR==FNR{a[$1]=$2;next}{name[FNR]=$1;split($2,z,"");for(i in z)seq[FNR][i]=z[i];if(length($1)>max1)max1=length($1);if(length($2)>max2)max2=length($2)}END{for(i=1;i<=max2;i+=width){printf("%"(max1+1)"s","");for(pos=i;pos<i+width&&pos<=max2;pos+=10)printf(pos-i>width-10?"%s":"%-10s",pos);print "";for(j in seq){printf("%"max1"s ",name[j]);for(k=i;k<=max2&&k<i+width;k++)printf("%s","\033[38;2;0;0;0m\033[48;2;"a[seq[j][k]]"m"seq[j][k]"\033[0m");print""}}}' "width=${1-60}" <(aacolor|sed $'s/ /\t/;s/ /;/g') -)
seqcol<temp
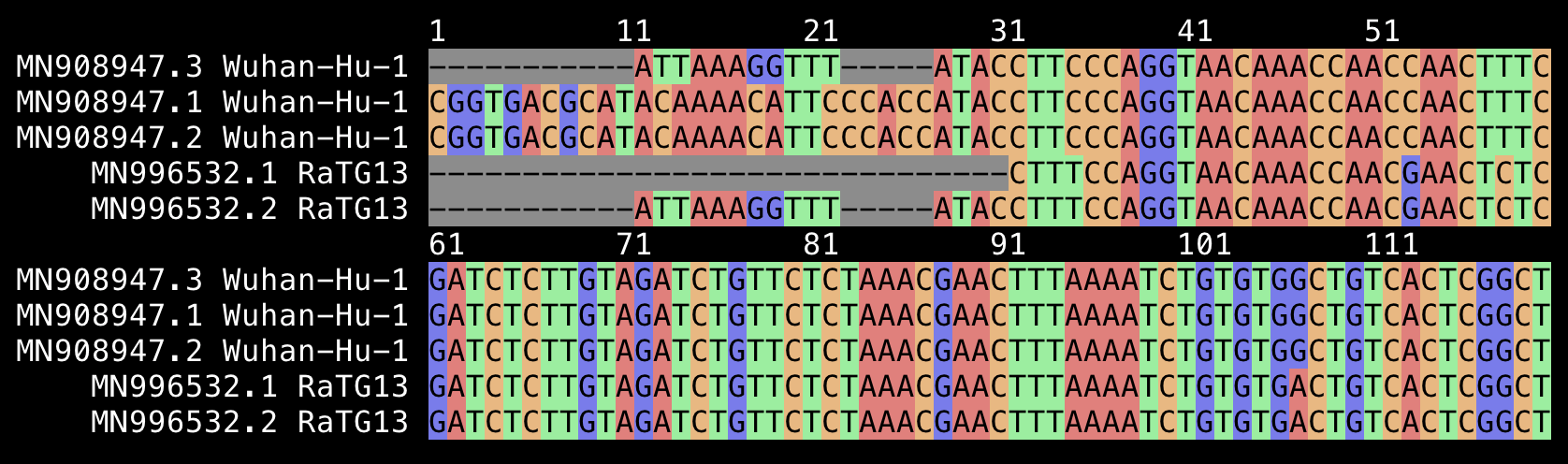
# make a TSV file for percentage identity between aligned sequences
curl -Ls sars2.net/f/pid.cpp>pid.cpp;g++ pid.cpp -O3 -o pid
curl -sL sars2.net/f/sarbe.fa.xz|gzip -dc>sarbe.fa
./pid<sarbe.fa>sarbe.pid
# matrix sort grep (in a TSV file with row names and column names, pick the first column whose
# name matches a substring and print the column sorted by its value along with the rownames)
masog()(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~x)break;next}{print$i,$1}' x="$1" "${2--}"|sort -n)
(masog ZC45 sarbe.pid|awk '$1>88{print$2}'|awk 'NR==FNR{a[$0];next}$0 in a' <(awk -F\\t 'NR>1{for(i=2;i<NR;i++)if($i>98&&$1!~/BANAL|ZC45|ZXC21/)next;print$1}' sarbe.pid|cut -d\ -f1);seqkit grep -nrp 'GD/P79-9/2019|BANAL|Tor2|MT121216.1' sarbe.fa|seqkit seq -ni)|seqkit grep -f- sarbe.fa|mafft --quiet --thread 4 ->nuc.fa;./pid<nuc.fa>nuc.pid
# install.packages("BiocManager");BiocManager::install("Biostrings")
library(ggplot2)
library(colorspace)
library(Biostrings)
t=readDNAStringSet("nuc.fa")
names(t)=sub("^.*? ","",sub(",.*","",names(t)))
gene=read.csv(header=F,text="ORF1a,266,13468
ORF1b,13468,21555
S,21563,25384
ORF3a,25393,26220
E,26245,26472
M,26523,27191
ORF6,27202,27387
ORF7a,27394,27759
ORF8,27894,28259
N,28274,29533
ORF10,29558,29674")
spike=read.csv(header=F,text="NTD,13,312
RBD,319,528
RBM,438,508
S1,1,686
S2,709,1273")
gene=rbind(cbind(spike[1],(spike[2]-1)*3+gene[3,2],(spike[3]-1)*3+2+gene[3,2]),gene)
m=as.matrix(t)
ref=m[grepl("Wuhan-Hu-1",rownames(m)),]
pos=seq_along(ref)-cumsum(ref=="-")
gene[,2]=sapply(gene[,2],\(x)which(pos==x)[1])
gene[,3]=sapply(gene[,3],\(x)which(pos==x)[1])
ranges=c(gene[1,3]+.5,gene[-1,2]-.5,gene[1,2],gene[nrow(gene),ncol(gene)])
targname="BtSY2"
targ=grep(targname,names(t),value=T)
step=1e3
heat=100*apply(gene[-1],1,\(x)as.matrix(stringDist(subseq(t,x[1],x[2]),"hamming"))[targ,]/(x[2]-x[1]+1))
# # don't count positions where either sequence has a gap
# heat=100*apply(gene[-1],1,\(x){block=ifelse(m=="-",NA,m)[,x[1]:x[2]];1-colMeans(block[targ,]==t(block),na.rm=T)})
colnames(heat)=gene[,1]
dist=100-read.table("nuc.pid",sep="\t",row.names=1,header=T)
hc=as.hclust(reorder(as.dendrogram(hclust(as.dist(dist))),cmdscale(dist,1)))
rownames(heat)=sub("Severe acute respiratory syndrome","SARS",rownames(heat))
rownames(heat)=sub(" (ORF|surface).*","",rownames(heat))
rownames(heat)=sub(" isolate "," ",rownames(heat))
pheatmap::pheatmap(heat,filename="0.png",legend=F,
clustering_callback=\(...)hc,cluster_cols=F,
gaps_col=nrow(spike),
cellwidth=17,cellheight=17,fontsize=9,treeheight_row=80,fontsize_number=8,treeheight_column=80,
border_color=NA,na_col="white",
display_numbers=round(heat),
number_color=matrix(ifelse(heat>.8*max(heat),"white","black"),nrow(heat))[hc$order,],
breaks=seq(0,max(heat),,256),
colorRampPalette(hex(HSV(c(210,210,210,160,110,60,40,20,0,0,0),c(0,.25,rep(.5,9)),c(rep(1,9),.5,0))))(256)
)
system(paste0("convert -gravity north \\( -splice 0x26 -pointsize 46 label:'Target: ",targname," (percentage of nucleotide changes)' \\) 0.png -append 1.png"))

curl -sL sars2.net/f/sarbe.fa.xz|gzip -dc>sarbe.fa
wget sars2.net/f/sarbe.pid
masog()(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i~x)break;next}{print$i,$1}' x="$1" "${2--}"|sort -n)
masog Hu-1 sarbe.pid|awk '$1>91{print$2}'|seqkit grep -f- sarbe.fa|mafft --thread 4 --quiet ->cum.fa
# install.packages("BiocManager");BiocManager::install("Biostrings")
library(ggplot2)
t=as.matrix(Biostrings::readAAStringSet("cum.fa"))
t[t=="n"]="-"
gene=read.csv(head=F,text="ORF1a,266,13468
ORF1b,13468,21555
S,21563,25384
ORF3a,25393,26220
E,26245,26472
M,26523,27191
ORF6,27202,27387
ORF7a,27394,27759
ORF8,27894,28259
N,28274,29533
ORF10,29558,29674")
geneh=c(1,1,1,1,2,3,4,1,2,3,4)
ref=t[grepl("Hu-1",rownames(t)),]
pos=1:length(ref)-cumsum(ref=="-")
gene[,2]=sapply(gene[,2],\(x)which(pos==x)[1])
gene[,3]=sapply(gene[,3],\(x)which(pos==x)[1])
t[,1:(gene[1,2]-1)]="N"
t[,gene[nrow(gene),3]:ncol(t)]="N"
genebars=c(gene[,2],gene[nrow(gene),3])
rownames(t)=sub("(,| ORF| orf| surface ).*","",rownames(t))
rownames(t)=sub("[^ ]* ","",rownames(t))
rownames(t)=sub(" isolate "," ",rownames(t))
rownames(t)=sub("Severe acute respiratory syndrome coronavirus 2 ","",rownames(t))
targ=rownames(t)[grep("Hu-1",rownames(t))]
m=apply(t(t[!rownames(t)==targ,])!=t[targ,],2,cumsum)
m=m[,order(-m[nrow(m),])]
xy=data.frame(x=1:nrow(m),y=c(m),z=rep(colnames(m),each=nrow(m)))
xy$z=factor(xy$z,unique(xy$z))
hue=c(0,210,100,60,300,160,25,250)+15
color=c("black",hcl(hue,120,60),"gray50",hcl(hue,120,30)[-c(6,7)],hcl(hue,40,80))
color=setNames(color[1:nrow(t)],rownames(t))[levels(xy$z)]
candidates=c(sapply(c(1,2,5),\(x)x*10^c(-10:10)))
xbreak=candidates[which.min(abs(candidates-max(xy$x)/10))]
ybreak=candidates[which.min(abs(candidates-(max(xy$y)-min(xy$y))/7))]
xend=max(xy$x)
ystart=ybreak*floor(min(xy$y,na.rm=T)/ybreak)
yend=ybreak*ceiling(max(xy$y,na.rm=T)/ybreak)
legend=data.frame(x=.02*xend,y=seq(yend*.9,,-yend/20,nlevels(xy$z)),label=levels(xy$z))
label=data.frame(label=gene[,1],x=rowMeans(gene[,-1]),y=yend-yend*(geneh-.2)/22)
ggplot(xy)+
geom_vline(xintercept=genebars,color="gray80",linewidth=.25)+
geom_vline(xintercept=c(0,xend),color="gray80",linewidth=.25)+
geom_hline(yintercept=c(ystart,yend),color="gray80",linewidth=.25)+
geom_line(aes(x,y,color=z),linewidth=.25)+
geom_label(data=legend,aes(x,y,label=label),color=color,size=2.2,hjust=0,fill=alpha("white",.85),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0)+
geom_label(data=label,aes(x=x,y=y,label=label),fill=alpha("white",.85),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0,size=2,hjust=.5)+
ggtitle(paste0("Cumulative mutations from ",targ," (UTRs masked)"))+
coord_cartesian(clip="off",expand=F)+
scale_x_continuous(limits=c(0,xend),breaks=seq(0,xend,xbreak),labels=\(x)ifelse(x>1e3,paste0(x/1e3,"k"),x),position="top")+
scale_y_continuous(limits=c(ystart,yend),breaks=seq(ystart,yend,ybreak),labels=\(x)ifelse(x>=1e3,paste0(x/1e3,"k"),x))+
scale_color_manual(values=color)+
theme(axis.text=element_text(size=6,color="black"),
axis.ticks=element_line(linewidth=.25,color="gray80"),
axis.ticks.length=unit(.2,"lines"),
axis.title=element_blank(),
legend.position="none",
panel.grid=element_blank(),
panel.background=element_rect(fill="white"),
plot.margin=margin(.5,.6,.5,.6,"lines"),
plot.title=element_text(size=8,color="black"))
ggsave("1.png",width=5,height=2.9,dpi=450)
If you plot the cumulative number of mutations between two sarbecoviruses, usually there is an increase in the slope of mutations at the start of the spike protein (because the NTD is more prone to mutations than the end of ORF1b).
But if SARS2 is compared to DB159, BtSY2, RaTG13, or BANAL-52, there is no increase in the slope, which suggests that there was a recombination event. ZC45, DB59, and BtSY2 are recombinants of SARS2-like and SARS1-like viruses, where the ORF1b is more similar to SARS1-like viruses. But then it's weird that there seems to be another recombination event in the S1, and the slope of cumulative mutations doesn't even change between the ORF1b and S1 (except in ZC45).
The version of the script below uses facets to make plots for each target sequence:
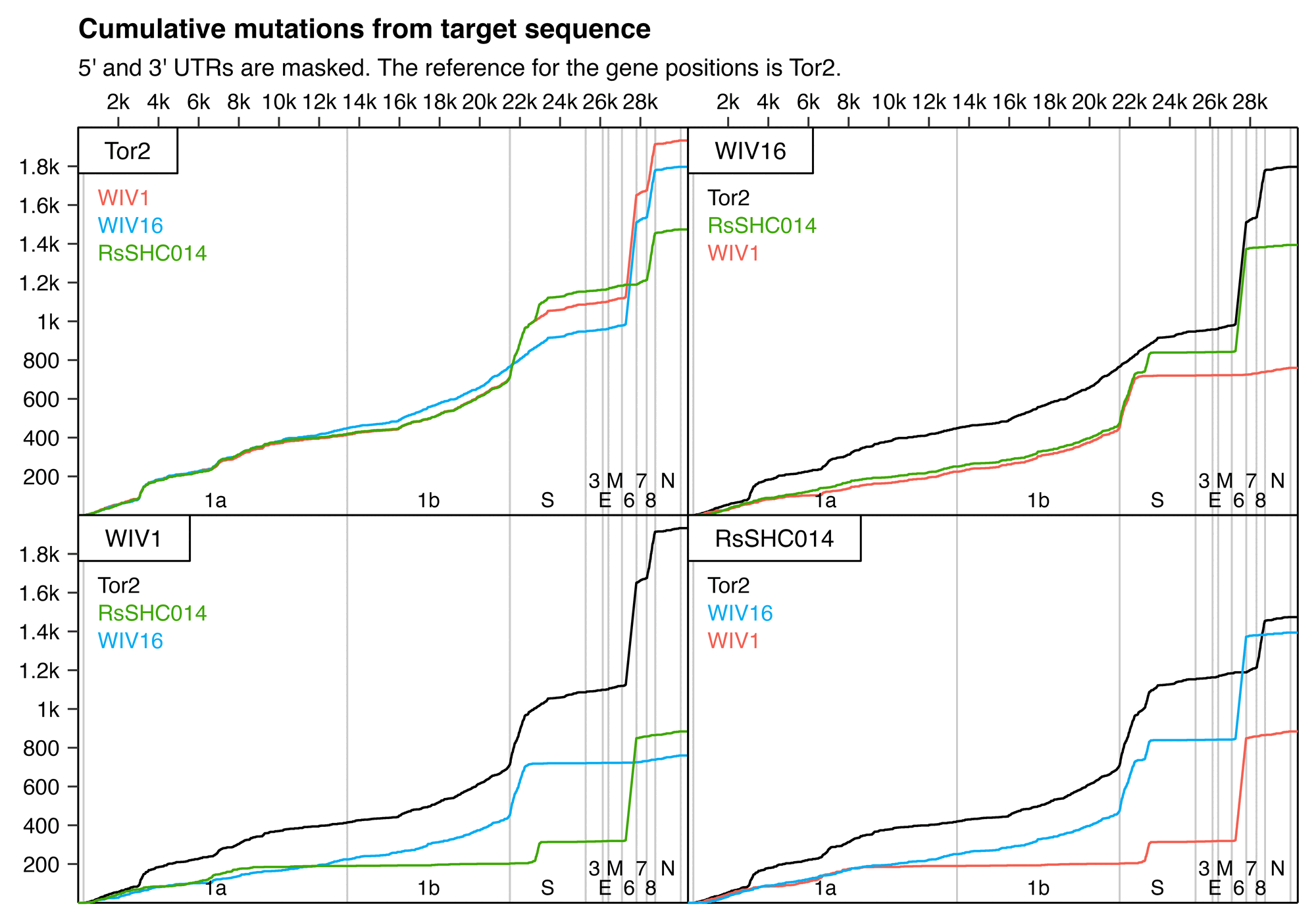
curl -sL sars2.net/f/sarbe.fa.xz|seqkit grep -nrp 'WIV1\b,WIV16,Tor2,RsSHC014'|mafft --quiet --thread 4 ->cum.fa
# install.packages("BiocManager");BiocManager::install("Biostrings")
library(ggplot2)
t=as.matrix(Biostrings::readAAStringSet("cum.fa"))
t[t=="n"]="-"
# Tor2
gene=read.csv(text="name,start,end,height
1a,265,13392,1
1b,13393,21485,1
S,21492,25259,1
3,25268,26153,2
E,26117,26347,1
M,26398,27063,2
6,27074,27265,1
7,27273,27772,2
8,27779,28118,1
N,28120,29388,2")
ref=t[grepl("Tor2",rownames(t)),]
pos=1:length(ref)-cumsum(ref=="-")
gene[,2]=sapply(gene[,2],\(x)which(pos==x)[1])
gene[,3]=sapply(gene[,3],\(x)which(pos==x)[1])
t[,1:(gene[1,2]-1)]="N"
t[,gene[nrow(gene),3]:ncol(t)]="N"
genebars=c(gene[,2],gene[nrow(gene),3])
rownames(t)=sub("(,| ORF| orf| surface ).*","",rownames(t))
rownames(t)=sub("[^ ]* ","",rownames(t))
rownames(t)=sub(" isolate "," ",rownames(t))
rownames(t)=sub("Severe acute respiratory syndrome coronavirus 2 ","",rownames(t))
rownames(t)=sub(".* ","",rownames(t))
p=do.call(rbind,lapply(1:nrow(t),\(i)data.frame(x=1:ncol(t),y=c(apply(t(t[-i,])!=t[i,],2,cumsum)),seq1=rownames(t)[i],seq2=rep(rownames(t)[-i],each=ncol(t)))))
lev=rownames(t)
# lev=c("Tor2","WIV1","WIV16","RsSHC014") # manually reorder panels
p$seq1=factor(p$seq1,lev);p$seq2=factor(p$seq2,lev)
hue=c(0,210,100,60,300,160,25,250)+15
color=c("black",hcl(hue,120,60),"gray50",hcl(hue,120,30)[-c(6,7)],hcl(hue,40,80))[1:nrow(t)]
cand=c(sapply(c(1,2,5),\(x)x*10^c(-10:10)))
xstep=cand[which.min(abs(cand-max(p$x)/10))]
ystep=cand[which.min(abs(cand-(max(p$y)-min(p$y))/7))]
xend=max(p$x)
ystart=ystep*floor(min(p$y,na.rm=T)/ystep)
yend=ystep*ceiling(max(p$y,na.rm=T)/ystep)
leg=p[p$x==max(p$x),];leg=leg[order(-leg$y),]
legend=do.call(rbind,lapply(split(leg,leg$seq1),\(x)data.frame(x=.03*xend,y=seq(yend*.82,,-yend/14,nrow(x)),seq1=x$seq1,seq2=x$seq2)))
label=data.frame(label=gene[,1],x=rowMeans(gene[,2:3]),y=yend*(gene$height-.2)/20)
ggplot(p)+
facet_wrap(~seq1,ncol=2,dir="v",strip.position="top")+
geom_vline(xintercept=genebars,color="gray80",linewidth=.25)+
geom_vline(xintercept=c(0,xend),linewidth=.25)+
geom_hline(yintercept=c(ystart,yend),linewidth=.25)+
geom_line(aes(x,y,color=seq2),linewidth=.3)+
geom_label(data=legend,aes(x,y,label=seq2,color=seq2),size=grid::convertUnit(unit(6,"pt"),"mm"),hjust=0,fill=alpha("white",.85),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0)+
geom_label(data=label,aes(x=x,y=y,label=label),fill=alpha("white",0),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0,size=2,hjust=.5)+
geom_label(data=subset(p,!duplicated(seq1)),aes(label=paste0("\n ",seq1," \n")),x=0,y=yend,lineheight=.5,hjust=0,vjust=1,size=2.3,label.r=unit(0,"lines"),label.padding=unit(0,"lines"),label.size=.25)+
labs(title="Cumulative mutations from target sequence",subtitle="5' and 3' UTRs are masked. The reference for the gene positions is Tor2.")+
scale_x_continuous(limits=c(0,xend),breaks=seq(xstep,xend-xstep*.8,xstep),labels=\(x)ifelse(x>1e3,paste0(x/1e3,"k"),x),position="top")+
scale_y_continuous(limits=c(ystart,yend),breaks=seq(ystep,yend-ystep,ystep),labels=\(x)ifelse(x>=1e3,paste0(x/1e3,"k"),x))+
scale_color_manual(values=color)+
coord_cartesian(clip="off",expand=F)+
theme(axis.text=element_text(size=6,color="black"),
axis.ticks=element_line(linewidth=.25),
axis.ticks.length=unit(.2,"lines"),
axis.title=element_blank(),
legend.position="none",
panel.spacing=unit(0,"pt"),
panel.background=element_blank(),
panel.grid=element_blank(),
plot.margin=margin(5,5,5,5),
plot.subtitle=element_text(size=6.5,margin=margin(0,,2)),
plot.title=element_text(size=7.5,face=2,margin=margin(0,,4)),
strip.background=element_blank(),
strip.text=element_blank())
ggsave("1.png",width=5,height=3.5,dpi=400*4)
system("mogrify -resize 25% 1.png")
# download sarbecovirus sequences
curl -sL sars2.net/f/sarbe.fa.xz|xz -dc>sarbe.fa
# efetch multi
emu()(curl -sd "id=$(paste -sd,)" "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1:-nuccore}&rettype=${2:-fasta}&retmode=$3")
# install SNAP
mkdir -p ~/bin;curl https://www.hiv.lanl.gov/repository/aids-db/PROGS/Snap/SNAP.pl>~/bin/snap;chmod +x ~/bin/snap
# run SNAP for a FASTA file converted to the format used by SNAP, where the ID is in the first column and the sequence is in the second column
sna()(~/bin/snap <(sed 's/ .*//' "$@"|seqkit fx2tab))
# convert the results of a SNAP run with two sequences to a simple CSV format
sna3()(sna "$@";(echo position,synonymous,nonsynonymous,indel;awk '/^[0-9]/{print$1,$7?$7:0,$8?$8:0,$2=="indel"}' OFS=, $(ls -t codons.*|head -n1))>snap.csv)
# codon align (align by translated amino acid sequence and insert gap triplets to nucleotide sequence)
coda()(x=$(cat);seqkit seq -g<<<"$x"|seqkit translate|mafft --quiet --thread 4 -|seqkit seq -s|gawk -F '' 'NR==FNR{gaps=0;for(i=1;i<=NF;i++)if($i=="-")a[NR][i-(gaps++)]++;next}{for(i=1;i<=NF;i+=3){i2=(i-1)/3+1;for(j=1;j<=a[FNR][i2];j++)printf"---";printf"%s",substr($0,i,3)};print""}' - <(seqkit seq -gs<<<"$x")|paste <(seqkit seq -n<<<"$x") -|seqkit tab2fx)
# download nucleotide coding sequences (fasta_cds_na)
seqkit grep -nrp Rs3367,RsSHC014 sarbe.fa|seqkit seq -ni|emu '' fasta_cds_na>temp
# see if example both ORF1ab and ORF1a are included so either of them has to be excluded
seqkit seq -n temp
# remove ORF1a, do codon alignment of concatenated CDS, and run SNAP
seqkit grep -nrvp '\b(1a|ORF1a)\b' temp|awk -F '_cds_' '!/>/||!a[$1]++'|coda|sna3
library(ggplot2)
# # Wuhan-Hu-1
# gene=read.csv(text="name,start,end,height
# 1a,266,13483,1
# 1b,13468,21555,1
# S,21563,25384,1
# 3a,25393,26220,1
# E,26245,26472,2
# M,26523,27191,3
# 6,27202,27387,1
# 7a,27394,27759,2
# 7b,27756,27887,3
# 8,27894,28259,1
# N,28274,29533,2
# 10,29558,29674,3")
# # Tor2
# gene=read.csv(text="name,start,end,height
# 1a,265,13392,1
# 1b,13393,21485,1
# S,21492,25259,1
# 3,25268,26153,2
# E,26117,26347,3
# M,26398,27063,1
# 6,27074,27265,2
# 7,27273,27772,3
# 8,27779,28118,1
# N,28120,29388,2")
# SHC014
gene=read.csv(text="name,start,end,height
1a,265,13413,1
1b,13398,21485,1
S,21492,25262,1
3a,25271,26095,1
3b,25692,26036,2
E,26120,26350,3
M,26401,27066,4
6,27077,27268,1
7a,27276,27644,2
7b,27641,27775,3
8,27782,28147,4
N,28162,29430,1")
gene$length=gene$end-gene$start+1
start=cumsum(c(0,gene$length/3))+1
gene$start=head(start,-1)
gene$end=start[-1]-1
t=read.csv("snap.csv")
xy=data.frame(x=t[,1],y=c(apply(t[-1],2,cumsum)),z=rep(colnames(t)[-1],each=nrow(t)))
xy$z=factor(xy$z,levels=unique(xy$z))
xstart=1;xend=nrow(t);ystart=0
cand=c(sapply(c(1,2,5),\(x)x*10^c(0:5)))
xstep=cand[which.min(abs(cand-max(xy$x)/8))]
ystep=cand[which.min(abs(cand-max(xy$y)/6))]
yend=ystep*ceiling(max(xy$y)*1.2/ystep)
ggplot(xy)+
geom_vline(color="gray70",xintercept=c(xstart,xend),linewidth=.2,lineend="square")+
geom_vline(color="gray70",xintercept=gene$start,linewidth=.2,lineend="square")+
geom_hline(color="gray70",yintercept=c(ystart,yend),linewidth=.2,lineend="square")+
geom_line(aes(x,y,color=z),linewidth=.4)+
geom_label(data=gene,hjust=.5,vjust=.5,aes(x=rowMeans(cbind(start,end)),label=name,y=yend-yend*(height-.5)*.06),size=2,fill=alpha("white",.85),label.r=unit(0,"lines"),label.padding=unit(.04,"lines"),label.size=0)+
labs(title="Concatenated CDS: Rs3367 vs RsSHC014",x=NULL,y=NULL)+
coord_cartesian(clip="off",expand=F)+
scale_x_continuous(limits=c(1,xend),breaks=seq(1,xend,xstep),position="top")+
scale_y_continuous(limits=c(0,yend),breaks=seq(0,yend,ystep))+
scale_color_manual(values=c("#00cc00","red","blue"))+
guides(colour=guide_legend(override.aes=list(linewidth=.4)))+
theme(axis.text=element_text(size=5.8,color="black"),
axis.ticks.length=unit(.15,"lines"),
axis.ticks=element_line(linewidth=.2,color="gray70"),
legend.background=element_blank(),
legend.key=element_rect(fill=alpha("white",0)),
legend.spacing.x=unit(.15,"lines"),
legend.key.size=unit(.8,"lines"),
legend.position="bottom",
legend.justification=c(1,0),
legend.margin=margin(-.7,0,-.2,0,"lines"),
legend.text=element_text(size=6.5,vjust=.5),
legend.title=element_blank(),
panel.background=element_rect(fill="white"),
panel.grid=element_blank(),
plot.margin=margin(.3,.4,.3,.3,"lines"),
plot.title=element_text(size=7,color="black",margin=margin(.1,0,.5,0,"lines")))
ggsave("1.png",width=3.3,height=2.3,dpi=450)
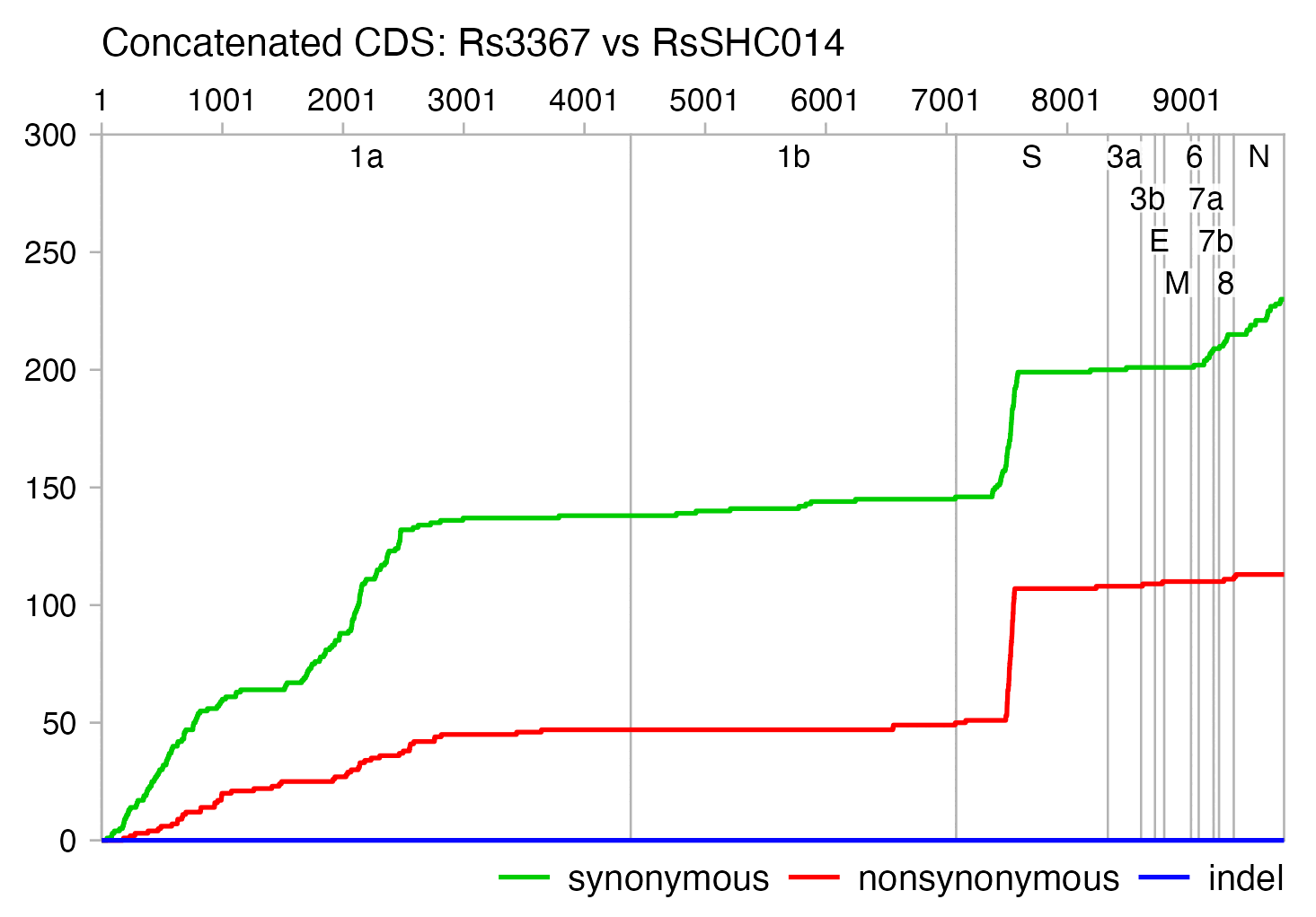
I'm using a Brazilian wastewater sample from November 2019 as an example (which was likely contaminated with a strain of SARS2 that was circulating in Brazil around March 2020):
enad()(printf %s\\n "${@-$(cat)}"|while IFS= read x;do curl -s "https://www.ebi.ac.uk/ena/portal/api/filereport?accession=$x&result=read_run&fields=fastq_ftp"|sed 1d|cut -f2|tr \; \\n;done|sed s,^,ftp://,|xargs wget -q)
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947'>sars2.fa
x=SRR13152143;enad $x
fastp -35 -i $x\_1.fastq.gz -I $x\_2.fastq.gz -o $x\_1.fq.gz -O $x\_2.fq.gz
minimap2 -a --sam-hit-only sars2.fa $x\_[12].fastq.gz|samtools sort ->$x.bam
This doesn't print the average quality at each position:
puptab0()(ruby -ane'next if$F[3]==0||$F[4]=="*";s=$F[4].upcase.gsub(/[.,]/,$F[2]).gsub(/\^./,"").gsub("$","");s2=s.dup;s.enum_for(:scan,/[+-]\d+/).each{m=Regexp.last_match;m.begin(0).upto(m.end(0)+s[m.begin(0)+1,m.end(0)].to_i-1){|i|s2[i]=" "}};s2.gsub!(" ","");t=s2.chars.tally;"ACGT".chars.map{|x|puts [$F[1],$F[2],x,t[x]||0,s2.length,"%.3f"%(100.0*(t[x]||0)/s2.length)]*" "}' "$@")
This also includes the average quality:
puptab()(ruby -ane's=$F[4].upcase.gsub(/[.,]/,$F[2]).gsub(/\^./,"").gsub("$","");s2=s.dup;s.enum_for(:scan,/[+-]\d+/).each{m=Regexp.last_match;m.begin(0).upto(m.end(0)+s[m.begin(0)+1,m.end(0)].to_i-1){|i|s2[i]=" "}};s2.gsub!(" ","");t=s2.chars.tally;"ACGT".chars.map{|x|q=$F[5].chars.values_at(*s2.chars.each_index.select{|i|s2[i]==x}).map{|c|c.ord-33};puts [$F[1],$F[2],x,t[x]||0,s2.size,"%.3f"%(100.0*(t[x]||0)/s2.size),q.size==0?"NA":"%.1f"%(q.sum.to_f/q.size)]*" "}' "$@")
Sample output:
$ samtools mpileup -f sars2.fa SRR13152144.bam 2>/dev/null|puptab|awk '$2!=$3'|(echo pos ref al count depth pct qual;cat)|head -n5|column -t pos ref al count depth pct qual 3392 G C 2 2 100.000 39.0 5706 A G 2 2 100.000 39.0 5929 T G 1 1 100.000 24.0 8576 A C 1 2 50.000 16.0
Another option is to use cpup, but it doesn't support
printing the average quality:
[https://github.com/y9c/cpup/blob/main/cpup.cpp]
$ wget https://github.com/y9c/cpup/raw/main/cpup.cpp $ g++ cpup.cpp -O3 --std=c++11 -o ~/bin/cpup $ samtools mpileup -f sars2.fa SRR13152144.bam|cpup|head -n3|column -t chr pos ref_base depth,A,C,G,T,N,Skip,Gap,Insert,Delete,,a,c,g,t,n,skip,gap,insert,delete MN908947.3 3357 A 2,1,0,0,0,0,0,0,0,0,1,0,0,0,0,0,0,0,0 MN908947.3 3358 T 2,0,0,0,1,0,0,0,0,0,0,0,0,1,0,0,0,0,0 $ samtools mpileup -f sars2.fa SRR13152144.bam|cpup -S|head -n3|column -t # -S (strandless) combines strands into one set of columns chr pos ref_base depth,a,c,g,t,n,skip,gap,insert,delete MN908947.3 3357 A 2,2,0,0,0,0,0,0,0,0 MN908947.3 3358 T 2,0,0,0,2,0,0,0,0,0
This thins out a FASTA file of sarbecoviruses to remove sequences with more than 99% identity to any previous sequence in the file, because otherwise for example there would be a huge number of different SARS1 sequences.
There's six different codons for arginine, but the rarest one is CGG which appears twice in the FCS insertion:
$ curl -sL sars2.net/f/sarbe.fa.xz|gzip -dc>sarbe.fa;wget sars2.net/f/sarbe.pid
$ thin()(awk -F\\t 'NR>1{for(i=2;i<NR;i++)if($i>x)next;print$1}' x="${1-99}" "${@:2}")
$ emu()(curl -sd "id=$(paste -sd,)" "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1:-nuccore}&rettype=${2:-fasta}&retmode=$3")
$ seqkit seq -ni sarbe.fa|emu '' fasta_cds_na>sarbe.na
$ thin<sarbe.pid|cut -d\ -f1|awk -F'lcl\\||_cds_' 'NR==FNR{a[$0];next}$2 in a' - <(seqkit fx2tab sarbe.na)|cut -f2|grep -o ...|egrep 'CG[ACGT]|AG[AG]'|LC_ALL=C sort|uniq -c|sort
1414 CGG
3560 CGA
8316 CGC
8649 AGG
17019 CGT
23149 AGA
One sequence:
$ curl -sL sars2.net/f/sarbe.fa.xz|gzip -dc>sarbe.fa
$ (seqkit grep -nrp Hu-1 sarbe.fa;seqkit grep -nrp Khosta-1 sarbe.fa)|mafft --thread 4 --quiet ->temp.fa
$ seqkit seq -s temp.fa|tr a-z A-Z|gawk -F '' 'NR==1{for(i=1;i<=length;i++){ref<a href=i-gaps[i]>i]=$i;if($i=="-")gap++;gaps[i]=gap}}NR==2{for(i=1;i<=length;i++)if($i!=ref[i])print ref[i</a>$i}'|grep -v -- -|head
G90A
G91T
C100A
C106T
T111C
T112C
C119T
C121T
A132G
T133C
Multiple sequences:
x=$(seqkit seq -u sarbe.fa|seqkit fx2tab);(grep Hu-1 <<<"$x";grep -v Hu-1 <<<"$x")|awk -F \\t 'NR==1{split($2,a,"");for(i=1;i<=length(a);i++){ref<a href=i-gaps[i]>i]=a[i];if(a[i]=="-")gap++;gaps[i]=gap};next}{split($2,a,"");o="";for(i=1;i<=length;i++)if(a[i]!=ref[i]&&a[i]!="-"&&ref[i]!="-")o=o","ref[i</a>a[i];print $1 FS substr(o,2)}'
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nucleotide&rettype=fasta&id=MN908947.3'>sars2.fa
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nucleotide&rettype=fasta_cds_aa&id=MN908947.3'>sars2.aa
$ seqkit seq -n sars2.aa|grep -v 'ORF1a\b'|sed 's/.*location=//;s/.*join(//'|tr , \\n|sed 's/[])].*//;s/\.\./ /'|while read l m;do seqkit subseq -r $l:$m sars2.fa|seqkit seq -s|sed s/^/$l\ /;done>temp
$ seqkit translate -l 1|sed 1,/Table/d|tr , \\n|tr -d \ |grep .|tr : \ |awk 'BEGIN{nt["A"];nt["C"];nt["G"];nt["T"]}NR==FNR{a[$1]=$2;next}{for(i=1;i<=length($2);i++){start=1+int((i-1)/3)*3;for(n in nt){if(n==substr($2,i,1))continue;alt=$2;alt=substr(alt,1,i-1)n substr(alt,i+1);if(a[substr($2,start,3)]==a[substr(alt,start,3)])print substr($2,i,1)i+$1-1 n}}}' - temp|head
G271A
C274T
T277A
T277C
T277G
C280A
C280T
C280G
T283A
T283C
(This erroneously treats C13468A as synonymous, because it's synonymous in ORF1a but not ORF1b.)
$ brew install bowtie2 seqkit samtools
[...]
$ wget ftp://ftp.ccb.jhu.edu/pub/data/bowtie_indexes/GRCh38_no_alt.zip;unzip GRCh38_no_alt.zip
[...]
$ seqkit seq -s sars2.fa|sed 's/A*$//'|gawk -F '' '{for(l=18;l<=30;l++)for(i=1;i<=length-l+1;i++)print ">"l"."i"\n"substr($0,i,l)}'>frag.fa
$ bowtie2 -p4 --no-unal -x GRCh38_noalt_as/GRCh38_noalt_as --score-min C,0,-1 -fU frag.fa>frag.sam
$ samtools view frag.sam|awk '{a[length($10)]++}END{for(i in a)print i,a[i]}' # number of matches for each fragment length
18 4915
19 1641
20 482
21 135
22 41
23 13
24 6
25 2
26 1
$ samtools view frag.sam|awk 'length($10)==26'|cut -f-10|tr \\t ' ' # longest match (26 bases)
26.7570 16 chr3 124017395 0 26M * 0 0 AGCCTTTACCTCCATTAGCATAGACA
--score-min C,0,-1 includes only exact matches.
You can download a FASTA file that contains 2-5 samples from different subtypes of HIV-1 and SIV sequences similar to HIV-1 from here: https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html. Set alignment type to "Subtype reference", change the region to "GENOME", and click "Get Alignment". Or alternatively you can download the file from here: f/HIV1_REF_2023_genome_DNA.fa.xz.
The following code slightly thins down the FASTA file so that it keeps only 50 random samples from the numeric subtypes, and then it makes a TSV percentage identity matrix for each pair of sequences in the file:
curl -Ls sars2.net/f/HIV1_REF_2023_genome_DNA.fa.xz|xz -dc>HIV1_REF_2023_genome_DNA.fa seqkit fx2tab HIV1_REF_2023_genome_DNA.fa|shuf|awk -F. '$2!~/^[0-9]/||++b<=50'|seqkit tab2fx>subref.fa curl -Ls sars2.net/f/pid.cpp>pid.cpp;g++ pid.cpp -O3 -o pid ./pid<subref.fa>subref.pid
And then this code plots the collection year on the x-axis and the distance to a specified target sequence on the y-axis:
library(ggplot2)
t=100-read.table("subref.pid",header=T,row.names=1)
rem=grepl("Ref......x",rownames(t))
t=t[!rem,!rem]
z=strsplit(rownames(t),"\\.")
year=as.numeric(sapply(z,"[",4))
year=year+ifelse(year<30,2000,1900)
name=paste0(sapply(z,"[",2),":",sapply(z,"[",3))
name=paste0(name,ifelse(grepl("CPZ|GOR",rownames(t)),paste0(":",sapply(z,"[",5)),""))
type=sapply(z,"[",2)
type[grepl("^\\d",type)]="Numeric subtypes"
type=sub("\\d+$","",type)
type=factor(type,unique(c("Numeric subtypes","CPZ","GOR",names(sort(tapply(year,type,mean))))))
p=data.frame(x=year,y=t[,grep("HXB2",rownames(t))])
seg=cbind(p[rep(1:nrow(p),each=2),],p[apply(t,1,\(x)order(x)[2:3]),])|>"colnames<-"(paste0("V",1:4))
ystart=0;yend=max(p$y)*1.05;xstart=min(p$x)-1;xend=max(p$x)+1
hue=c(0,220,120,60,30,300)+15
color=c("black","gray60","gray30",hcl(hue,100,60),hcl(hue,100,25)[-5],hcl(hue,50,80))
ggplot(p,aes(x,y))+
# geom_segment(data=seg,aes(x=V1,y=V2,xend=V3,yend=V4),color="gray50",linewidth=.15)+
ggforce::geom_mark_hull(aes(color=type,fill=type),concavity=1000,radius=unit(.15,"cm"),expand=unit(.15,"cm"),alpha=.13,size=.15)+
geom_point(aes(color=type),size=.3)+
# geom_text(aes(color=type),label=name,size=2,vjust=-.7,show.legend=F)+
ggrepel::geom_text_repel(aes(color=type),label=name,size=1.9,max.overlaps=Inf,segment.size=.1,min.segment.length=.2,box.padding=.07,show.legend=F)+
coord_cartesian(clip="off",expand=F)+
scale_x_continuous(limits=c(xstart,xend),breaks=seq(1980,2020,5))+
scale_y_continuous(limits=c(ystart,yend),breaks=seq(0,yend,5),labels=\(x)paste0(x,"%"))+
labs(x=NULL,y="Distance to SIVcpzMB66",title="Nucleotide distance percent in whole genome to to SIVcpzMB66 (closest SIV to early subtype B)",subtitle="Positions where either sequence has a gap are ignored. Source: hiv.lanl.gov/content/sequence/NEWALIGN/align.html, \"subtype reference\" collection. Only 50 random samples from numeric subtypes displayed."|>stringr::str_wrap(92))+
scale_color_manual(values=color)+
scale_fill_manual(values=color)+
guides(color=guide_legend(nrow=2,override.aes=list(linewidth=0,size=.5,fill=NA)))+
theme(axis.text=element_text(size=6.5,color="black"),
axis.ticks=element_line(linewidth=.2,color="gray50"),
axis.ticks.length=unit(.15,"lines"),
axis.title=element_text(size=8),
legend.key=element_rect(fill=alpha("white",0)),
legend.spacing=unit(0,"lines"),
legend.key.size=unit(.7,"lines"),
legend.key.width=unit(1.1,"lines"),
legend.box.spacing=unit(0,"pt"),
legend.position="bottom",
legend.justification=c(.5,.5),
legend.margin=margin(.3,0,0,0,"lines"),
legend.text=element_text(size=7.2,vjust=.5),
legend.title=element_blank(),
panel.border=element_rect(color="gray50",fill=NA,linewidth=.2),
panel.grid=element_blank(),
panel.background=element_rect(fill="white"),
plot.title=element_text(size=8.1,margin=margin(0,0,.6,0,"lines")),
plot.subtitle=element_text(size=7,margin=margin(0,0,.6,0,"lines")))
ggsave("1.png",width=5.2,height=3.5,dpi=450)
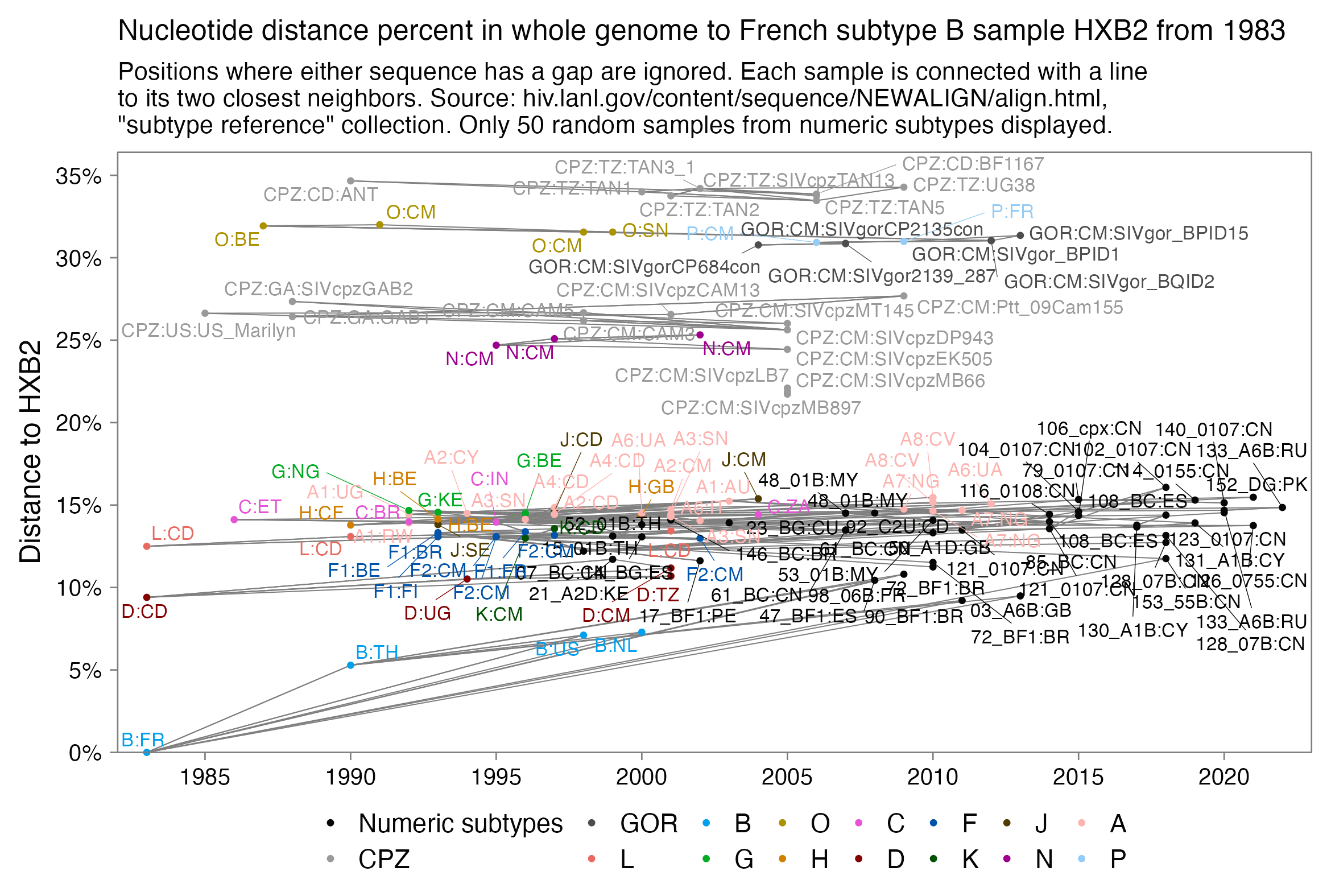
I'm using the Los Alamos HIV subtype reference as an example, which also contains HIV-1-like SIV sequences from chimpanzees and gorillas: https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html. I picked only the oldest sample from each single-letter subtype along with all the SIV sequences:
brew install iqtree2
curl -Ls sars2.net/f/HIV1_REF_2023_genome_DNA.fa.xz|xz -dc>HIV1_REF_2023_genome_DNA.fa
seqkit fx2tab HIV1_REF_2023_genome_DNA.fa|awk -F. '{print ($4>50?1900:2000)+$4,$0}'|sort -n|cut -d\ -f2-|awk -F. '($2~/^[A-Z][0-9]?$/&&!a[$2]++)||/CPZ|GOR|\.[ONP]\./'|seqkit tab2fx|sed 's/Ref.\(.*\)\.[^.]*/\1/'>small5.fa
iqtree2 -nt auto -fast -s small5.fa
-nt auto sets the number of threads automatically
instead of using a single thread.
The tree is unrooted but here I rerooted the tree so that I picked the sample that was the furthest from HXB2 as the outgroup:
library(ggtree);library(ape)
t=read.tree("small5.fa.treefile")
t=root(t,"CPZ.CD.90.ANT",resolve.root=T)
ggtree(t)+geom_tiplab()+xlim(0,2)
ggsave("1.png",height=length(t$tip.label)*.15,width=5)
system("mogrify -trim -bordercolor white -border 32 1.png")
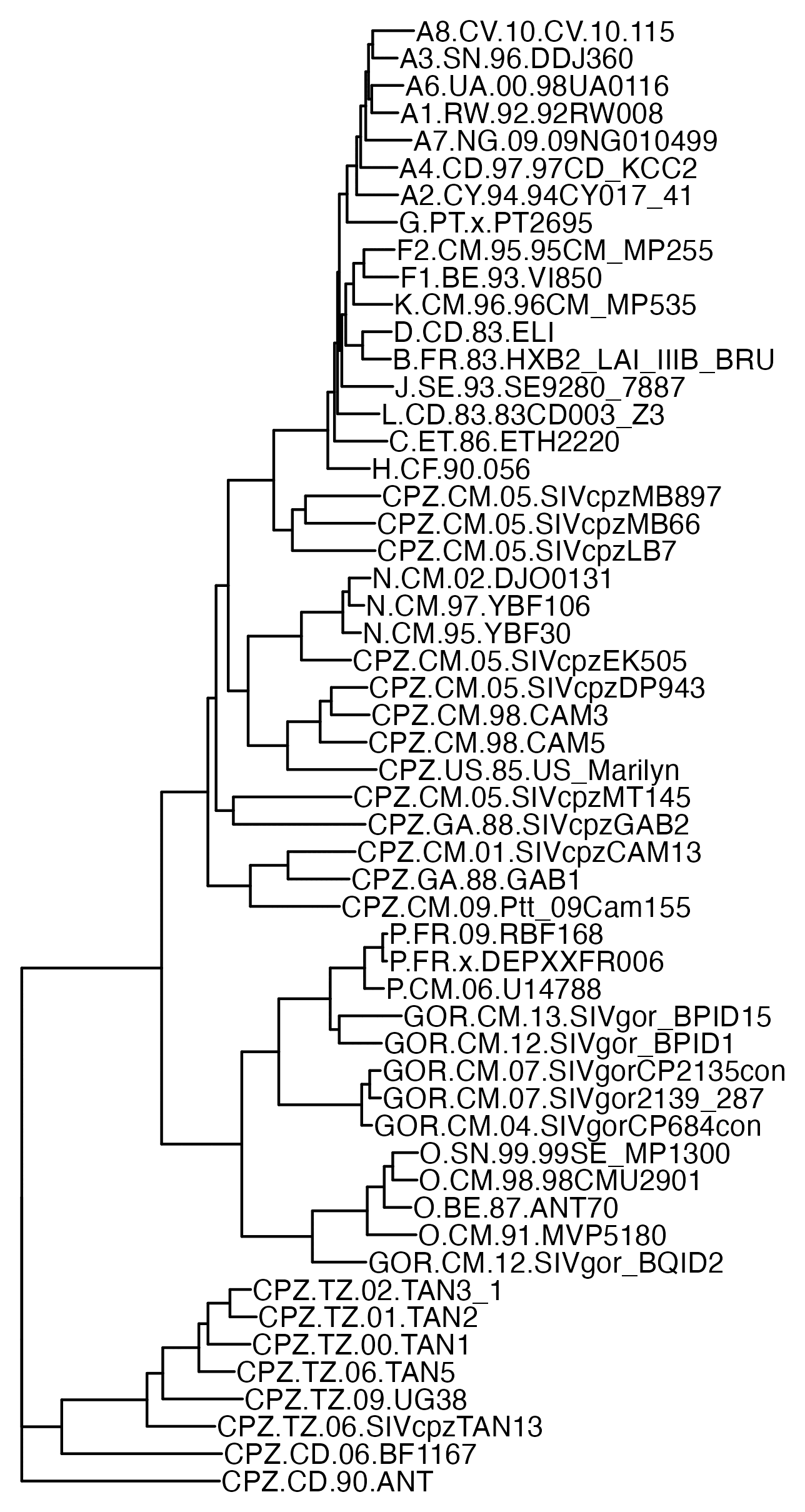
# download sarbecovirus sequences
curl -sL sars2.net/f/sarbe.fa.xz|xz -dc>sarbe.fa
# efetch multi (download each accession from STDIN)
emu()(curl -sd "id=$(paste -sd,)" "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=${1:-nuccore}&rettype=${2:-fasta}&retmode=$3")
# download nucleotide coding sequences of sarbecoviruses
seqkit seq -ni sarbe.fa|emu '' fasta_cds_na>sarbe.na
# install SNAP
mkdir -p ~/bin;curl https://www.hiv.lanl.gov/repository/aids-db/PROGS/Snap/SNAP.pl>~/bin/snap;chmod +x ~/bin/snap
# compile C++ program for making a percentage identity matrix
curl -Ls sars2.net/f/pid.cpp>pid.cpp;g++ pid.cpp -O3 -o ~/bin/pid
# run SNAP for a FASTA file converted to the format used by SNAP, where the ID is in the first column and the sequence is in the second column
sna()(~/bin/snap <(sed 's/ .*//' "$@"|seqkit fx2tab))
# run SNAP and make a space-delimited file for syn and nonsyn mutations in each pair of sequences
sna2()(sna "$@";awk '/This is comparison/{x=$7;y=$8}/^[0-9]/{print x,y,$1,$7?$7:0,$8?$8:0}' $(ls -t codons.*|head -n1)>snap.tab)
# codon align (align by translated amino acid sequence and insert gap triplets to nucleotide sequence)
coda()(x=$(cat);seqkit seq -g<<<"$x"|seqkit translate|mafft --quiet --thread 4 -|seqkit seq -s|gawk -F '' 'NR==FNR{gaps=0;for(i=1;i<=NF;i++)if($i=="-")a[NR][i-(gaps++)]++;next}{for(i=1;i<=NF;i+=3){i2=(i-1)/3+1;for(j=1;j<=a[FNR][i2];j++)printf"---";printf"%s",substr($0,i,3)};print""}' - <(seqkit seq -gs<<<"$x")|paste <(seqkit seq -n<<<"$x") -|seqkit tab2fx)
# rename FASTA defline to use shorter names
shorn()(sed -E $'s/(,| surface| ORF1| orf1| genomic sequence).*//;s/Severe acute respiratory syndrome/SARS/;s/ isolate / /')
# pick 24 random spike sequences and rename the sequences to include the sequence names on the defline (SARS1, sequences with N letters, and sequences shorter than 3700 bases are excluded)
seqkit grep -nrp gene=S,spike,surface sarbe.na|seqkit seq -m3700|seqkit grep -svrp N|sed 's/lcl|//;s/_cds_.*//'|seqkit fx2tab|awk -F\\t 'NR==FNR{a[$1]=$2;next}{print">"$1" "a[$1]"\n"$2}' <(seqkit seq -n sarbe.fa|sed $'s/ /\t/') -|seqkit grep -nrvp'SARS coronavirus'|shorn|seqkit shuffle|seqkit head -n24|coda>snap.fa
# run SNAP and make percentage identity matrix
sna2 snap.fa;~/bin/pid<snap.fa>snap.pid
t=data.table::fread("snap.tab") # this is much faster for large files than read.csv
t=t[,.(V4=sum(V4),V5=sum(V5)),.(V1,V2)] # V4 is syn, V5 is nonsyn, V1 is seq1, V2 is seq2
t=rbind(t,t[,.(V1=V2,V2=V1,V4,V5)]) # add flipped pairs for each pair of compared sequences
m=t[,xtabs(V4/V5~V1+V2)]
name=sub("^>","",grep("^>",readLines("snap.fa"),value=T))
name=setNames(sub("^.*? ","",name),sub(" .*","",name))
id=rownames(m)
# rownames(m)=colnames(m)==paste0(name[rownames(m)]," (",sub("\\.\\d+$","",rownames(m)),")") # include accessions
rownames(m)=colnames(m)=name[rownames(m)] # don't include accessions
dist=read.table("snap.pid",header=T,row.names=1,sep="\t")
rownames(dist)=colnames(dist)=sub(" .*","",rownames(dist))
dist=dist[id,id]
# reorder clustering tree based on first dimension in MDS to arrange similar branches together
hc=as.hclust(reorder(as.dendrogram(hclust(dist(dist))),cmdscale(dist(dist),1)))
disp=ifelse(m>=10,round(m),sprintf("%.1f",m));disp[m==0]=0
m=m^.6 # use curved scale similar to weak log scale for colors
m[is.infinite(m)]=NA # replace ratios where nonsyn is zero with NA because pheatmap doesn't support infinite values
maxcolor=max(m,na.rm=T)
pal=hsv(c(21,21,21,16,11,6,3,0)/36,c(0,.25,rep(.5,6)),c(rep(1,8)))
pheatmap::pheatmap(m,filename="1.png",display_numbers=disp,clustering_callback=\(...)hc,
legend=F,cellwidth=13,cellheight=13,fontsize=9,fontsize_number=8,
border_color=NA,na_col="gray90",number_color="black",
breaks=seq(0,maxcolor,,256),colorRampPalette(pal)(256))
tit="Ratio of synonymous to nonsynonymous mutations in sarbecovirus spike sequences"
sub="Calculated with SNAP.pl: hiv.lanl.gov/content/sequence/SNAP/help_files/README.html. The ratio is an unadjusted ratio of raw mutation counts, and not a dS/dN ratio with Jukes-Cantor correction. The clustering tree is based on a nucleotide percentage identity matrix of spike sequences."
system(paste0("f=1.png;mar=20;w=`identify -format %w $f`;magick \\( -size $[w-mar*2]x -font Arial-Bold -pointsize 45 caption:'",gsub("'","'\\''",tit),"' -splice $[mar]x20 \\) \\( -size $[w-mar*2]x -font Arial -pointsize 40 caption:'",gsub("'","'\\''",sub),"' -splice $[mar]x8 \\) \\( $f -splice x6 \\) -append 1.png"))