RFK Jr. wrote the following on his Substack: [https://
New York Post reporter Jon Levine got it wrong in his article today claiming that I said COVID was "ethnically targeted" to spare Jews.
I have never, ever suggested that the COVID-19 virus was targeted to "spare" Jews. I accurately pointed out - during an off-the-record conversation - that China and other governments are developing ethnically targeted bioweapons and that a 2021 study of the COVID-19 virus shows that COVID-19 appears to disproportionately affect certain races since the furin cleave docking site and least compatible with ethnic Chinese, Finns, and Ashkenazi Jews.
In that sense, it serves as a kind of proof of concept for ethnically targeted bioweapons. I do not believe and never implied that the ethnic effect was deliberately engineered.
That study is here: https://
pubmed. .ncbi. nlm. nih. gov/ 32664879/
The paper RFK linked was actually from 2020 and not 2021, and it
looked at mutations in the ACE2 and TMPRSS2 genes and not the furin
gene, even though TMPRSS2 also has a role in the cleavage of the spike
protein by furin: [https://
For SARS-CoV-2 to enter cells, its surface glycoprotein spike (S) must be cleaved at two different sites by host cell proteases, which therefore represent potential drug targets. In the present study, we show that S can be cleaved by the proprotein convertase furin at the S1/S2 site and the transmembrane serine protease 2 (TMPRSS2) at the S2' site. We demonstrate that TMPRSS2 is essential for activation of SARS-CoV-2 S in Calu-3 human airway epithelial cells through antisense-mediated knockdown of TMPRSS2 expression. Furthermore, SARS-CoV-2 replication was also strongly inhibited by the synthetic furin inhibitor MI-1851 in human airway cells. In contrast, inhibition of endosomal cathepsins by E64d did not affect virus replication. Combining various TMPRSS2 inhibitors with furin inhibitor MI-1851 produced more potent antiviral activity against SARS-CoV-2 than an equimolar amount of any single serine protease inhibitor.
But anyway, in the paper that RFK linked on his Substack, there were
only two references to Ashkenazis, which were in the caption of figure 1
and in the following part of text which talked about the same figure:
[https://
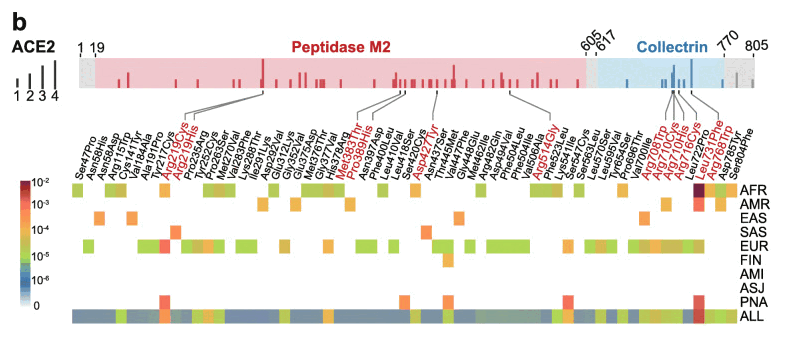
Specifically, 39% (24/61) and 54% (33/61) of deleterious variants in ACE2 occur in African/African-American (AFR) and Non-Finnish European (EUR) populations, respectively (Fig. 1b). Prevalence of deleterious variants among Latino/Admixed American (AMR), East Asian (EAS), Finnish (FIN), and South Asian (SAS) populations is 2-10%, while Amish (AMI) and Ashkenazi Jewish (ASJ) populations do not appear to carry such variants in ACE2 coding regions (Fig. 1b).
Figure 1b shows that the number of deleterious ACE2 alleleles which occurred at least once was 0 in Ashkenazis and Amishes, 1 in Finns, 2 in South Asians, and so on:
However there's a bias where populations with a larger sample size
are more likely to have one or more occurrence of a deleterious allele
than populations with a smaller sample size. In gnomAD v3 which was used
in the paper, some populations have a much smaller population size than
other populations, so for example the number of samples that have been
typed for the K26R allele of ACE2 is 53,215 for the population labeled
"European (non-Finnish)" but only 2,650 for Ashkenazis
and 684 for Amishes. [https://
You can see the frequency of ACE2 alleles in gnomAD v3 from here:
https://
>al=strsplit( " Ser47Pro Asn58His Asn58Asp Arg115Trp Cys141Tyr Val184Ala Ala191Pro Tyr217Cys Arg219Cys Arg219His Pro235Arg Tyr252Cys Pro263Ser Met270Val Val283Phe Lys288Thr Ile291Lys Asp292Val Glu312Lys Gly352Val Glu375Asp Met376Thr Gly377Val His378Arg Met383Thr Pro389His Asn397Asp Phe400Leu Leu410Val Leu418Ser Ser420Cys Asp427Tyr Asn437Ser Thr445Met Val447Phe Gly448Glu Met462Ile Arg482Gln Asp494Val Phe504Leu Phe504Ile Val506Ala Arg514Gly Phe523Leu Lys541Ile Ser547Cys Ser563Leu Leu570Ser Leu595Val Tyr654Ser Pro696Thr Val700Ile Arg708Trp Arg710Cys Arg710His Arg716Cys Leu722Pro Leu731Phe Arg768Trp Asp785Tyr Ser804Phe", " ")[[ 1]] > t=read. csv( " gnomAD_ v3. 1. 2_ ENSG00000130234_ 2023_ 07_ 18_ 20_ 18_ 13. csv", check. names=F) > rows=na. omit( match( paste0( " p. ", al), t$` HGVS Consequence`)) > total=colSums( t[ rows, grepl( " Allele Number ", colnames( t))]) > deleterious=colSums( t[ rows, grepl( " Allele Count ", colnames( t))]) > o=data. frame( row. names=sub( " Allele Number ", " ", names( total)), deleterious, total, ratio=deleterious/ total) > o=o[ order( o$ ratio),]; o$ ratio=sprintf( "%. 7f", o$ ratio); o deleterious total ratio Amish 0 41769 0. 0000000 Middle Eastern 0 14554 0. 0000000 Ashkenazi Jewish 0 161388 0. 0000000 European (Finnish) 1 367360 0. 0000027 South Asian 3 162912 0. 0000184 East Asian 5 218231 0. 0000229 Latino/ Admixed American 19 640622 0. 0000297 European (non- Finnish) 136 3241463 0. 0000420 Other 6 91784 0. 0000654 African/ African American 455 1873327 0. 0002429
So basically the output above shows that the total number of deleterious alleles is so miniscule that it won't make much difference.
If you look at TMPRSS2 instead of ACE2, the total number of alleles that were classified as deleterious is about 2 orders of magnitude bigger, and the ratio of deleterious alleles is the lowest in Ashkenazis and the highest in Finns:
>al=strsplit( " Gly6Arg Tyr20Cys Glu23Ala Tyr37Cys Pro54Leu Thr58Met Leu91Gln Leu91Pro Gly142Arg Gly142Trp Asp144Glu Glu145Lys Cys148Phe Arg150Leu Val160Met Val171Met Gly181Arg Gly189Ala Gly189Cys Tyr190Asp Cys231Ser Leu239Phe Arg240Cys Arg255Ser Val257Met Gly259Ser Ala262Val Trp267Arg Gly282Arg Ile286Phe Thr287Pro Val292Met Ala295Gly Cys297Ser Val298Met Tyr322Cys His334Leu Pro335Leu Ser339Phe Ala347Glu Ala347Thr Ala347Val Phe357Ser Val364Ala Val364Leu Gly370Ser Gly383Arg Trp384Leu Thr387Ala Gly391Glu Ile405Thr Met424Val Gly432Ala Gly432Glu Asp435Tyr Gln438Glu Pro444Leu Gly457Arg Ser460Arg Gly462Asp Gly462Ser Cys465Tyr Arg470Ile", " ")[[ 1]] > t=read. csv( " gnomAD_ v3. 1. 2_ ENSG00000184012_ 2023_ 07_ 18_ 23_ 46_ 03. csv", check. names=F) > rows=na. omit( match( paste0( " p. ", al), t$` HGVS Consequence`)) > total=colSums( t[ rows, grepl( " Allele Number ", colnames( t))]) > deleterious=colSums( t[ rows, grepl( " Allele Count ", colnames( t))]) > o=data. frame( row. names=sub( " Allele Number ", " ", names( total)), deleterious, total, ratio=deleterious/ total) > o=o[ order( o$ ratio),]; o$ ratio=sprintf( "%. 7f", o$ ratio); o deleterious total ratio Ashkenazi Jewish 493 214382 0. 0022996 Latino/ Admixed American 2436 943212 0. 0025827 Middle Eastern 56 19498 0. 0028721 Other 430 128910 0. 0033357 European (non- Finnish) 15547 4200196 0. 0037015 South Asian 1157 297238 0. 0038925 Amish 234 56286 0. 0041573 African/ African American 12223 2557968 0. 0047784 East Asian 2007 320878 0. 0062547 European (Finnish) 4172 653440 0. 0063847
However almost all of the difference in the number of deleterious TMPRSS2 alleles is accounted by Val160Met, which has a minor allele frequency ranging from about 14% in Ashkenazis to about 39% in Finns:
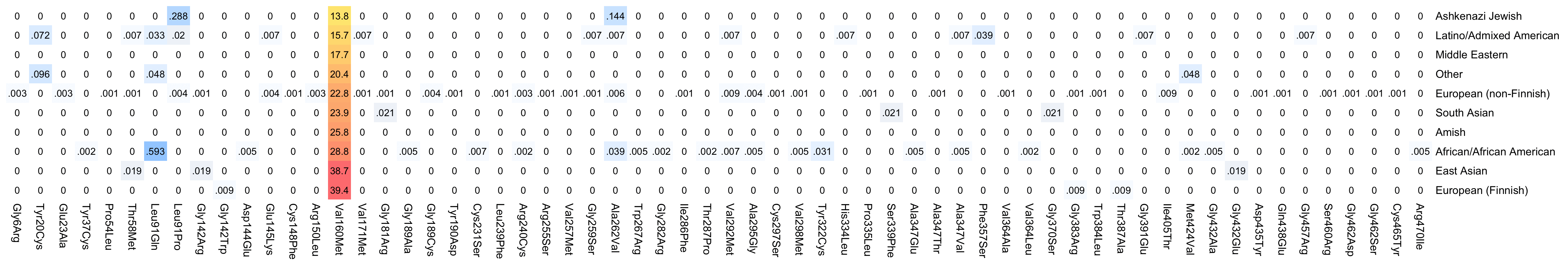
So if the Val160Met allele does not actually have that much impact on suspectibility to COVID, then Ashkenazis may not have a significant advantage in terms of their profile of TMPRSS2 alleles either. And actually if Val160Met is excluded, then Ashkenazis have the second-highest ratio of deleterious TMPRSS2 alleles in gnomAD v3:
deleterious total ratio Amish 0 55378 0.0000000 Middle Eastern 0 19182 0. 0000000 European (Finnish) 3 642848 0. 0000047 East Asian 3 315700 0. 0000095 South Asian 3 292408 0. 0000103 European (non- Finnish) 61 4132214 0. 0000148 Other 4 126818 0. 0000315 Latino/ Admixed American 35 927936 0. 0000377 Ashkenazi Jewish 15 210912 0. 0000711 African/ African American 304 2516608 0. 0001208
Another paper which has been used as a source for the claim that
Ashkenazis are less suspectible to COVID than other ethnic groups is a
paper by Ali et al. from 2020 titled "ACE2 coding
variants in different populations and their potential impact on
SARS-CoV-2 binding affinity". [https://
The part of the paper that conspiratards focused on was the figure shown below, which lists the 6 alleles that were analyzed in the paper so that they are ordered by their modeled level of electrostatic interaction with the spike protein of SARS-CoV-2. The K26R allele which is the most common in Ashkenazis has a red background because it's the only allele which was modeled as beneficial, but the other 5 alleles were modeled as detrimental so they have a green background:

In the figure above, East Asians are shown to have the highest frequency of the I468V allele, which was modeled as the least harmful of the 5 deleterious alleles, and which some people thought meant that East Asians would have the second-most advantageous mutation profile after Ashkenazis. But actually even though the I468V allele is modeled as only mildly deleterious in the paper, and even though only about 1.1% of East Asians have the allele at gnomAD v2, the other deleterious alleles analyzed in the paper are also so rare that if you calculate a weighted average of the frequency of each allele at gnomAD multiplied by the modeled interaction energy of each mutated form of ACE2, then East Asians end up having the lowest total interaction energy level, which might theoretically make them the most suspectible to COVID. But all 6 alleles listed in the paper are so rare that there's only a tiny range of variation in the weighted averages of the energy levels, so that they range from about -40.222 kcal/mol in East Asians to about -40.175 kcal/mol in Ashkenazis:
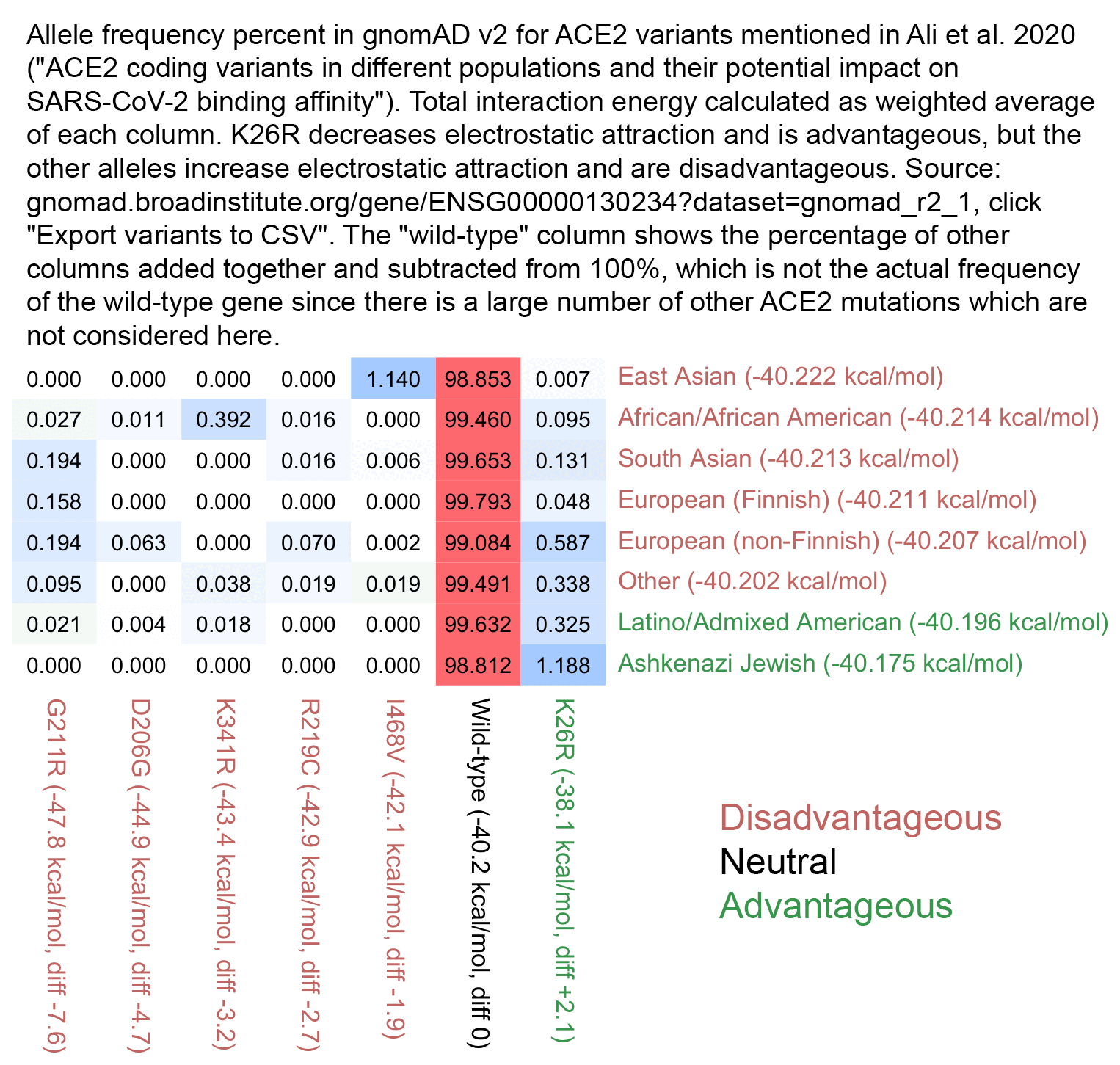
Here's R code to reproduce the plot above:
#install. #packages( " BiocManager") BiocManager:: #install( " ComplexHeatmap") install. #packages( " circlize" install. library(packages( " colorspace") ComplexHeatmap) library( circlize) # for colorRamp2 library(colorspace) native=- 40. 2 energy=read. csv( header=F, text=" G211R,- 47. 8 D206G,- 44. 9 K341R,- 43. 4 R219C,- 42. 9 I468V,- 42. 1 K26R,- 38. 1") codon=read. csv( header=F, row. names=1, text=" A, Ala C, Cys D, Asp E, Glu F, Phe G, Gly H, His I, Ile K, Lys L, Leu M, Met N, Asn P, Pro Q, Gln R, Arg S, Ser T, Thr V, Val W, Trp X, Ter Y, Tyr") # freq=read. freq=read.csv( " gnomAD_ v2. 1. 1_ ENSG00000130234_ 2023_ 07_ 18_ 15_ 15_ 32. csv", check. names=F) # go to https:// gnomad. broadinstitute. org/ gene/ ENSG00000130234? dataset=gnomad_ r2_ 1 and click " Export variants to CSV" csv( " https:// pastebin. com/ raw/ sqKQ7Lk7", check. names=F) name=paste0( " p. ", codon[ substr( energy[, 1], 1, 1),], sub( ".(.*). ", "\\ 1", energy[, 1]), codon[ sub( ".*(.) ", "\\ 1", energy[, 1]),]) rows=match( name, freq$` HGVS Consequence`) freq1=freq[ rows, grepl( " Allele Number ", colnames( freq))] freq2=freq[ rows, grepl( " Allele Count ", colnames( freq))] diff=round( energy[, 2]- native, 1) diff=ifelse( diff> 0, paste0( "+ ", diff), diff) m=freq2/ freq1 sums=( 1- colSums( m))* native+ colSums( m* energy[, 2]) rownames( m) =paste0( energy[, 1], " (", energy[, 2], " kcal/ mol, diff ", diff, ") ") colnames( m) =paste0( sub( " Allele Number ", " ", colnames( freq1)), " (", sprintf( "%. 3f", sums), " kcal/ mol) ") m=m[, order( sums)] m=rbind( m, 1- colSums( m)) rownames( m)[ nrow( m)] =" Wild type (- 40. 2 kcal/ mol, diff 0) " m=m[ c( 1: 5, 7, 6),] m=t( m)* 100 disp=apply( m, 2, sprintf, fmt="%. 3f") m=sqrt( m) m[ is. na( m)] =0 colcol=hcl( c( 0, 0, 0, 0, 0, 0, 120)+ 15, c( 60, 60, 60, 60, 60, 0, 60), c( 60, 60, 60, 60, 60, 0, 60)) rowcol=hcl( c( 0, 0, 0, 0, 0, 0, 120, 120)+ 15, 60, 60) maxcolor=max( m) png( " 1. png", w=ncol( m)* 60+ 2000, h=nrow(