In the 370-base partial RdRp sequence of 4991/RaTG13 which was
published in 2016, there's 5 nucleotide changes and 1 amino acid change
from Wuhan-Hu-1. So it has about 98.6% nucleotide identity with
Wuhan-Hu-1 even though the whole genome of RaTG13 only has 96.1%
identity, so people like Monali Rahalkar have been speculating that the
rest of the genome of RaTG13 was fabricated to make it appear to be more
distant to SARS2: [https://
WIV had deposited RdRp and ORF8 submission of 4991 in 2018 and so when they sequenced sars2 they should have found 4991 RdRp of almost 3000 bases with 98 percent and protein match should be 99 or more. What's the guarantee that the rest was not that close too! Did they distance it
Was this the reason Shi lost her sleep that 4991 RdRp was 98 percent and on aa much more closer to sars2. So little tampering of this sequence into a synthetic or serial passage could result in a lab made sars2, and people would suspect them?
When the RaTG13 sequence came out on 27 Jan 2020, it was about 96.2 percent similar. Did they adjust it at other places so that the average came low? It's like 98, 99 percent marks in many subjects and S protein 93 lowering the average marks to 96.2%?
However in the paper by Ge et al. from 2016 where the RdRp fragment
was published, it was specifically called the "conserved 440-bp RdRp fragment": "Only two sequences detected in this study were homologous
to betacoronaviruses. One of them (RaBtCoV/4991) was detected in a R.
affinis sample and was related to SL-CoV. The conserved 440-bp RdRp
fragment of RaBt-CoV/4991 had 89% nt identity and 95% aa identity with
SL-CoV Rs672 (Yuan et al., 2010)."
[https://
The same region was also sequenced in the "discovery of a rich gene pool" paper by Hu et
al. from 2016: "A total of 602 alimentary specimens
(anal swabs or feces) were collected and
tested for the presence of CoVs by a Pan-CoV RT-PCR targeting the 440-nt
RdRp fragment that is conserved among all known α- and β-CoVs [20]." [https://
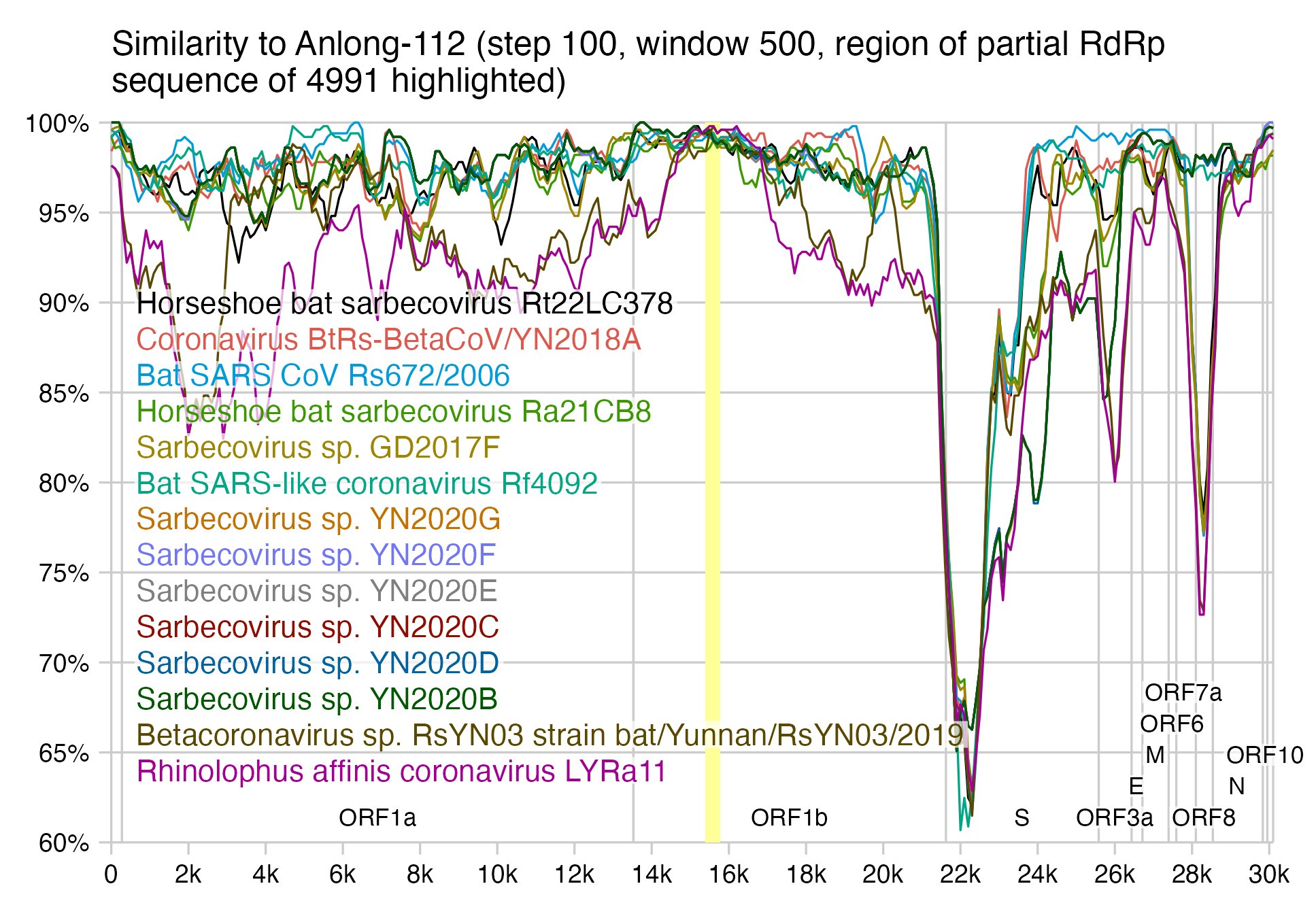
In the region of the RdRp fragment of RaTG13, for example Tor2 has only 3 nucleotide changes from WIV1 and SHC014, even though it has about 93-95% identity to them in the whole genome (but here the identity is a bit higher because I ignored positions where either sequence had a gap and I did a big multiple sequence alignment). Here the first column shows the number of nucleotide changes in the region of the partial RdRp sequence of 4991, and the second column shows the identity percentage in the whole genome:
$curl -sL sars2. net/ f/ sarbe. fa. xz| gzip -dc> sarbe. fa; seqkit seq -g sarbe. fa> sarbe. ungap $ wget sars2. net/ f/ sarbe. pid $ bow()([[ -e $1. 1. bt2 ]]|| bowtie2- build -- threads 3 "$ 1" "$ 1"; bowtie2 -- no- unal -p3 -x "$@ ") $ curl ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=fasta& id=KP876546' > 4991. fa $ bow sarbe. ungap -fU 4991. fa -a -- score- min L,- 1. 5,- 1. 5| grep -v ^@| ruby -ane' puts [$ F[ 2],$ F[ 3],$ F[ 3]. to_ i+$ F[ 5]. scan(/\ d+(? =[ MD])/). map( &: to_ i). sum- 1]* " " '| while read l m n; do seqkit grep -p $l sarbe. ungap| seqkit subseq -r $m:$ n; done| seqkit grep -nrvp ' SARS coronavirus ZJ01'| mafft -- quiet -- thread 4 -> temp $ x=Tor2; cat <( seqkit grep -nrp $x temp) temp| sed ' s/\(,\| surface\).*// '| seqkit fx2tab| awk -F\\ t ' NR==1{ split($ 2, a, " "); next}{ split($ 2, b, " "); d=0; for( i=1; i< =length; i++) if( a[ i]! =b[ i]) d++; print d,$ 1} '| sort -n| awk ' NR==FNR{ a[$ 2] =$ 1; next}{$ 1=$ 1"\ t" a[$ 2]} 1' <( awk -F\\ t ' NR==1{ for( i=2; i< =NF; i++) if($ i~ x) break; next}{ print$ i,$ 1} ' x="$ x" sarbe. pid) -| sed ' s/ /\ t/ '| awk '$ 2==100||($ 2< 98& &$ 0!~/ SARS coronavirus/) '| head| column -ts$ '\ t' 0 100. 0000 NC_ 004718. 3 SARS coronavirus Tor2 1 93. 5641 KP886808. 1 Bat SARS- like coronavirus YNLF_ 31C 1 93. 5506 KP886809. 1 Bat SARS- like coronavirus YNLF_ 34C 2 92. 0783 KU973692. 1 UNVERIFIED: Severe acute respiratory syndrome- related coronavirus isolate F46 2 93. 9236 KY417148. 1 Bat SARS- like coronavirus isolate Rs4247 3 95. 4596 KC881005. 1 Bat SARS- like coronavirus RsSHC014 3 95. 7228 KC881006. 1 Bat SARS- like coronavirus Rs3367 3 95. 6781 KF367457. 1 Bat SARS- like coronavirus WIV1 3 95. 8298 KY417144. 1 Bat SARS- like coronavirus isolate Rs4084 3 93. 8966 KY417149. 1 Bat SARS- like coronavirus isolate Rs4255
RhGB05 and RhGB06 have only 1 nucleotide change in the region, but they have about 97% identity in the whole genome.
For example Anlong-112 also has 1-3 nucleotide changes in the same region with all the viruses in this simplot, even though the identity in the whole genome is around 90-96%:
Rahalkar also posted a screenshot of a SARS2 sample from 2024 at
BLAST which had only 3 nucleotide changes from the 370-base RdRp
fragment of 4991, and she wrote: "So now the virus
is mutating back to the ancestor! Because the three base diff is a
recent most sequence." [https://
$curl ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=fasta& id=KP876546' > 4991. fa $ curl https:// data. nextstrain. org/ files/ ncov/ open/ global/ aligned. fasta. xz| gzip -dc> global. fa $ seqkit locate -f4991. fa -m8 <( seqkit head -n1 global. fa)| sed 1d| cut -f5, 6| seqkit subseq -r $( tr \\ t :) global. fa| seqkit grep -nrp ' 2024$ '| seqkit grep -svp N| cat 4991. fa -| seqkit seq -s| awk ' NR==1{ split($ 0, a, " "); l=length; next}{ split($ 0, b, " "); d=0; for( i=1; i< =l; i++) if( a[ i]! =b[ i]) d++; n[ d]++} END{ for( i in n) print i, n[ i]} ' 4 1 5 390 6 56 7 3
Limeng Yan was making a big deal out of how the envelope protein of
ZC45 had 100% amino acid identity to SARS2: [https://
The E protein is tolerant of mutations as evidenced in both SARS (Figure 2A) and related bat coronaviruses (Figure 2B). This tolerance to amino acid mutations of the E protein is further evidenced in the current SARS-CoV-2 pandemic. After only a short two-month spread of the virus since its outbreak in humans, the E proteins in SARS-CoV-2 have already undergone mutational changes. Sequence data obtained during the month of April reveals that mutations have occurred at four different locations in different strains (Figure 2C). Consistent with this finding, sequence blast analysis indicates that, with the exception of SARS-CoV-2, no known coronaviruses share 100% amino acid sequence identity on the E protein with ZC45/ZXC21 (suspicious coronaviruses published after the start of the current pandemic are excluded[18,27-31]), Although 100% identity on the E protein has been observed between SARS-CoV and certain SARS-related bat coronaviruses, none of those pairs simultaneously share over 83% identity on the Orf8 protein[32]. Therefore, the 94.2% identity on the Orf8 protein, 100% identity on the E protein, and the overall genomic/amino acid-level resemblance between SARS-CoV-2 and ZC45/ZXC21 are highly unusual. Such evidence, when considered together, is consistent with a hypothesis that the SARS-CoV-2 genome has an origin based on the use of ZC45/ZXC21 as a backbone and/or template for genetic gain-of-function modifications.
But if SARS1 has an identical envelope protein on the amino acid level with viruses where the ORF8 is less than 83% identical, then it suggests that there's nothing unusual if there's 100% aa identity for the envelope proteins of viruses that are not closely related.
The envelope protein of BtKY72 also has 100% amino acid identity to other African sarbecoviruses even though they only have about 85% nucleotide identity in the whole genome:
$curl -sL sars2. net/ f/ sarbe. fa. xz| gzip -dc> sarbe. fa; wget sars2. net/ f/ sarbe. pid $ emu()( curl -sd " id=$( paste -sd,) " " https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=${ 1:- nuccore} & rettype=${ 2:- fasta} & retmode=$ 3") $ seqkit seq -ni sarbe. fa| emu ' ' fasta_ cds_ aa| seqkit grep -nrp ' gene=E\ b| envelope| protein=E\ b' > env. aa $ sed ' s/ lcl|//; s/_ prot.*// ' env. aa| seqkit fx2tab| awk -F\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ print" > "$ 1" " a[$ 1] "\ n"$ 2} ' <( seqkit seq -n sarbe. fa| sed $' s/ /\ t/; s/\(,\| surface\| ORF1\).*// ') -> env2. aa $ masog()( awk -F\\ t ' NR==1{ for( i=2; i< =NF; i++) if($ i~ x) break; next}{ print$ i,$ 1} ' x="$ 1" "${ 2--} "| sort -n) $ pid0()( seqkit fx2tab "$@ "| awk -F\\ t ' NR==1{ split($ 2, a, " "); l=length; next}{ split($ 2, b, " "); d=0; n=0; for( i=1; i< =l; i++){ n++; if( a[ i]! =b[ i]) d++} print 100*( 1- d/ n),$ 1} ') $ x=BtKY72; seqkit grep -nrp $x env2. aa| cat - env2. aa| pid0| sort -rn| awk ' NR==FNR{ a[$ 2] =$ 1; next}{$ 1=$ 1" " a[$ 2]} 1' <( masog $x sarbe. pid) -| head 100 85. 3537 MT726045. 1 Sarbecovirus sp. isolate PREDICT/ PRD- 0038/ AABSMF 100 85. 4618 MT726044. 1 Bat coronavirus isolate PREDICT/ PDF- 2370/ OTBA35RSV 100 85. 4618 MT726043. 1 Sarbecovirus sp. isolate PREDICT/ PDF- 2386/ OTBA40RSV 100 100. 0000 KY352407. 1 Severe acute respiratory syndrome- related coronavirus strain BtKY72 99. 359 81. 7080 MZ190137. 1 Bat SARS- like coronavirus Khosta- 1 strain BtCoV/ Khosta- 1/ Rh/ Russia/ 2020 97. 4359 78. 1437 OR233314. 1 Horseshoe bat sarbecovirus isolate Rt22SL92 97. 4359 78. 1471 OR233313. 1 Horseshoe bat sarbecovirus isolate Rt22SL85 97. 4359 78. 1471 OR233312. 1 Horseshoe bat sarbecovirus isolate Rt22SL58 97. 4359 78. 1437 OR233311. 1 Horseshoe bat sarbecovirus isolate Rt22SL130 97. 4359 78. 0657 OR233303. 1 Horseshoe bat sarbecovirus isolate Rt22SL9
Tor2 and HKU3 also have an identical envelope protein on the amino acid level even though they only have about 88% nucleotide identity in the whole genome (in a multiple sequence alignment where positions where either sequence has a gap are ignored):
$x=Tor2; seqkit grep -nrp $x env2. aa| cat - env2. aa| pid0| sort -rn| awk ' NR==FNR{ a[$ 2] =$ 1; next}{$ 1=$ 1" " a[$ 2]} 1' <( masog $x sarbe. pid) -| awk '$ 1==100'| sort -nk2| head 100 82. 2274 OR233309. 1 Horseshoe bat sarbecovirus isolate Rt22SL67 100 88. 0533 DQ084199. 1 bat SARS coronavirus HKU3- 2 100 88. 0562 DQ084200. 1 bat SARS coronavirus HKU3- 3 100 88. 0597 DQ022305. 2 Bat SARS coronavirus HKU3- 1 100 88. 1371 OK017837. 1 Sarbecovirus sp. isolate JX2021C 100 88. 2051 OK017801. 1 Sarbecovirus sp. isolate HB2020D 100 88. 2333 OK017845. 1 Sarbecovirus sp. isolate JX2021P 100 88. 2441 OK017808. 1 Sarbecovirus sp. isolate FJ2021A 100 88. 3719 OK017844. 1 Sarbecovirus sp. isolate JX2021O 100 88. 3878 OQ503499. 1 Severe acute respiratory syndrome- related coronavirus isolate RsHB190371
The paper by DRASTIC titled "Unexpected novel
Merbecovirus discoveries in agricultural sequencing datasets from Wuhan,
China" said: [https://
A significant amount of reads of a SARS-r CoV most closely related to HKU3 was identified in a Mus Musculus splenocyte RNA-seq sequencing dataset SRR5819071 in BioProject PRJNA393936. The novel bat SARS-like HKU3-related CoV was identified via BLAST analysis of the dataset, with the reads only 96-98% similar to their closest related GenBank sequences (Supp. Fig. 27, Supp. Table 4). Although the sequences are far too few to be assembled, the presence of transcription regulatory sequence (TRS)-leader associated fusion reads between the TRS-leader and the TRS-N gene suggest active replication of the HKU3-r CoV within the Mus Musculus splenocyte sample.
Even though most of the other runs discussed in the paper were submitted by Huazhong Agricultural University or Fujian Agriculture and Forestry University, the SRR5819071 run which contained the HKU3-like virus was submitted by WIV.
When I aligned the reads in the run against sarbecovirus sequences, I got the highest number of covered bases for the isolates 141376 and 141341 which were published in 2023, and the average error rates for them were only about 0.40% and 0.08%:
$curl -sL sars2. net/ f/ sarbe. fa. xz| gzip -dc> sarbe. fa $ seqkit seq -g sarbe. fa| seqkit fx2tab| sed $' s/ A*\ t$// '| seqkit tab2fx| bbmask. sh window=20 in=stdin out=sarbe. mask $ bowtie2- build -- threads 3 sarbe. fa{,} $ x=SRR5819071; bowtie2 -p 3 -- no- unal -x sarbe. mask -1 ${ x}_ 1. fastq. gz -2 ${ x}_ 2. fastq. gz -- score- min L,- 1,- 1| samtools sort -@ 3 ->$ x. sarbe. bam $ covera()( samtools coverage "$ 1"| awk \$ 4| cut -f1, 3- 6|( gsed -u ' 1s/$/\ terr%\ tname/; q'; sort -rnk5| awk -F\\ t -v OFS=\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ print$ 0, sprintf( "%. 2f", a[$ 1])} ' <( samtools view $x| awk -F\\ t '{ x=$ 3; n[ x]++; len[ x]+ =length($ 10); sub(/.* NM: i:/, " "); mis[ x]+ =$ 1} END{ for( i in n) print i"\ t" 100* mis[ i]/ len[ i]} ') -| awk -F\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ print$ 0"\ t" a[$ 1]} ' <( seqkit seq -n "${ 2-~/ viral. fa} "| gsed ' s/ /\ t/; s/,.*// ') -)| column -ts$ '\ t') $ covera $x. sarbe. bam sarbe. mask # rname endpos numreads covbases coverage err% name OQ503496. 1 29646 10 1355 4. 5706 0. 40 Severe acute respiratory syndrome- related coronavirus isolate 141376 OQ503495. 1 29657 8 837 2. 82227 0. 08 Severe acute respiratory syndrome- related coronavirus isolate 141341 OQ297705. 1 30029 846586 350 1. 16554 6. 01 MAG: Severe acute respiratory syndrome- related coronavirus isolate CQ/ L80. 18/ 2021 OP776339. 1 30600 3 234 0. 764706 15. 93 Sarbecovirus sp. isolate RhGB05 OQ297732. 1 29568 2 200 0. 676407 1. 33 MAG: Severe acute respiratory syndrome- related coronavirus isolate GD/ L105. 18/ 2022 OK017815. 1 29550 2 193 0. 65313 0. 33 Sarbecovirus sp. isolate GD2017I KF294457. 1 29676 2 182 0. 61329 0. 00 Bat SARS- like coronavirus isolate Longquan- 140 orf1ab polyprotein OL674076. 1 29780 116980 159 0. 533915 20. 76 Severe acute respiratory syndrome- related coronavirus isolate Rs7907 OK017841. 1 29350 1 150 0. 511073 0. 67 Sarbecovirus sp. isolate JX2021L OQ503500. 1 30385 146 151 0. 496956 21. 70 Severe acute respiratory syndrome- related coronavirus isolate 4105 OQ503498. 1 30172 2 137 0. 454063 16. 33 Severe acute respiratory syndrome- related coronavirus isolate BDA190366 KY417144. 1 29770 2 132 0. 443399 0. 00 Bat SARS- like coronavirus isolate Rs4084 OK017842. 1 29337 2 120 0. 40904 0. 00 Sarbecovirus sp. isolate JX2021M OQ503504. 1 30325 11539 118 0. 389118 22. 24 Severe acute respiratory syndrome- related coronavirus isolate 162173
When I ran MEGAHIT on the reads which aligned against sarbecoviruses, my longest contig was 535 bases long and it was 100% identical to both 141376 and 141341:
x=SRR5819071;samtools view $x. sarbe. bam| grep -Ev ' OQ297705| OL674076| OQ503504'| cut -f1, 10| seqkit tab2fx> temp. fa megahit -r temp. fa -- min- count 1 -o megatemp; seqkit sort -lr megatemp/ final. contigs. fa
The 141376 and 141341 isolates actually have over 5% nucleotide distance from HKU3:
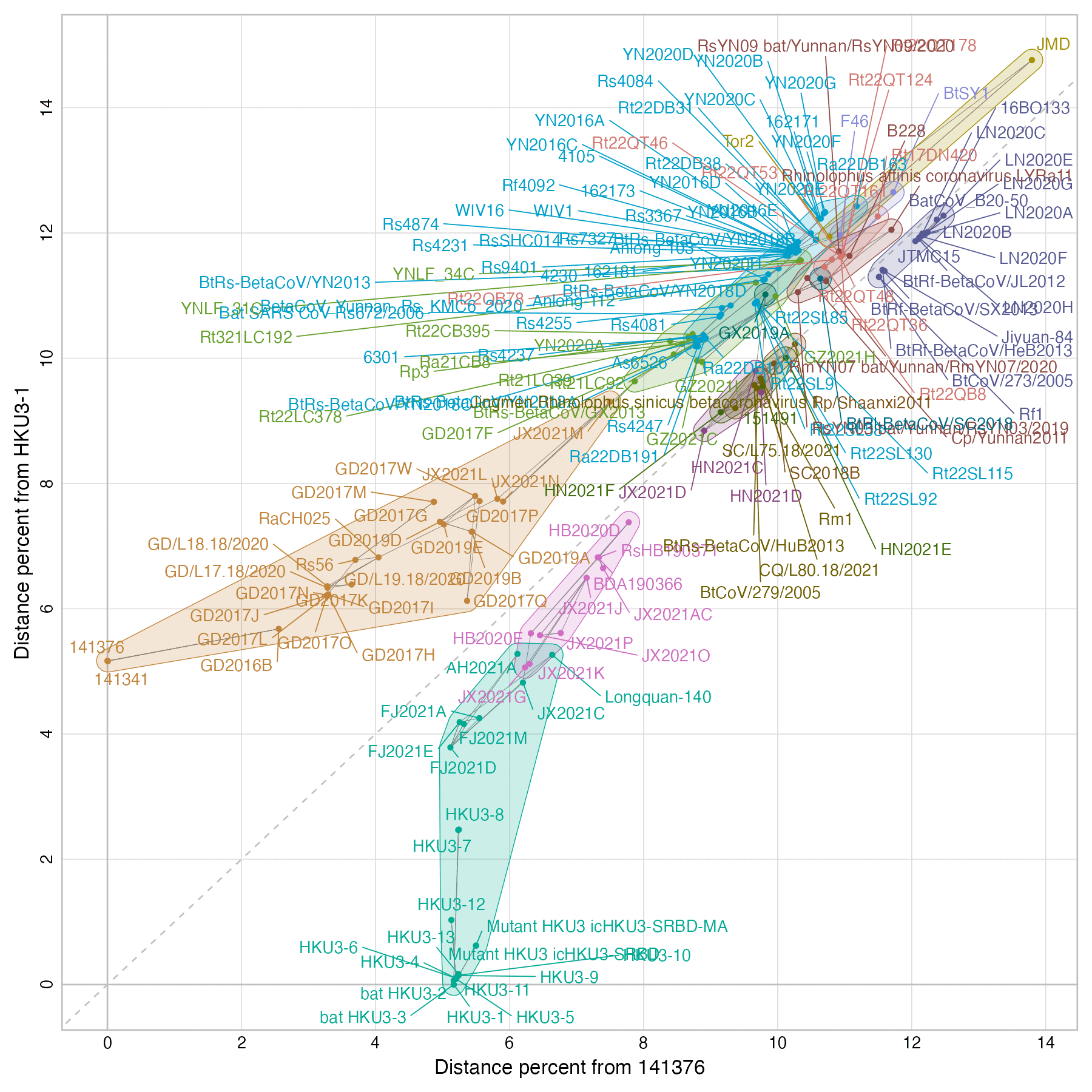
The 141376 and 141341 isolates were published in a paper by WIV
titled "Isolation of ACE2-dependent and -independent
sarbecoviruses from Chinese horseshoe bats", where the
corresponding authors were Zhengli Shi and Michael Letko. [https://
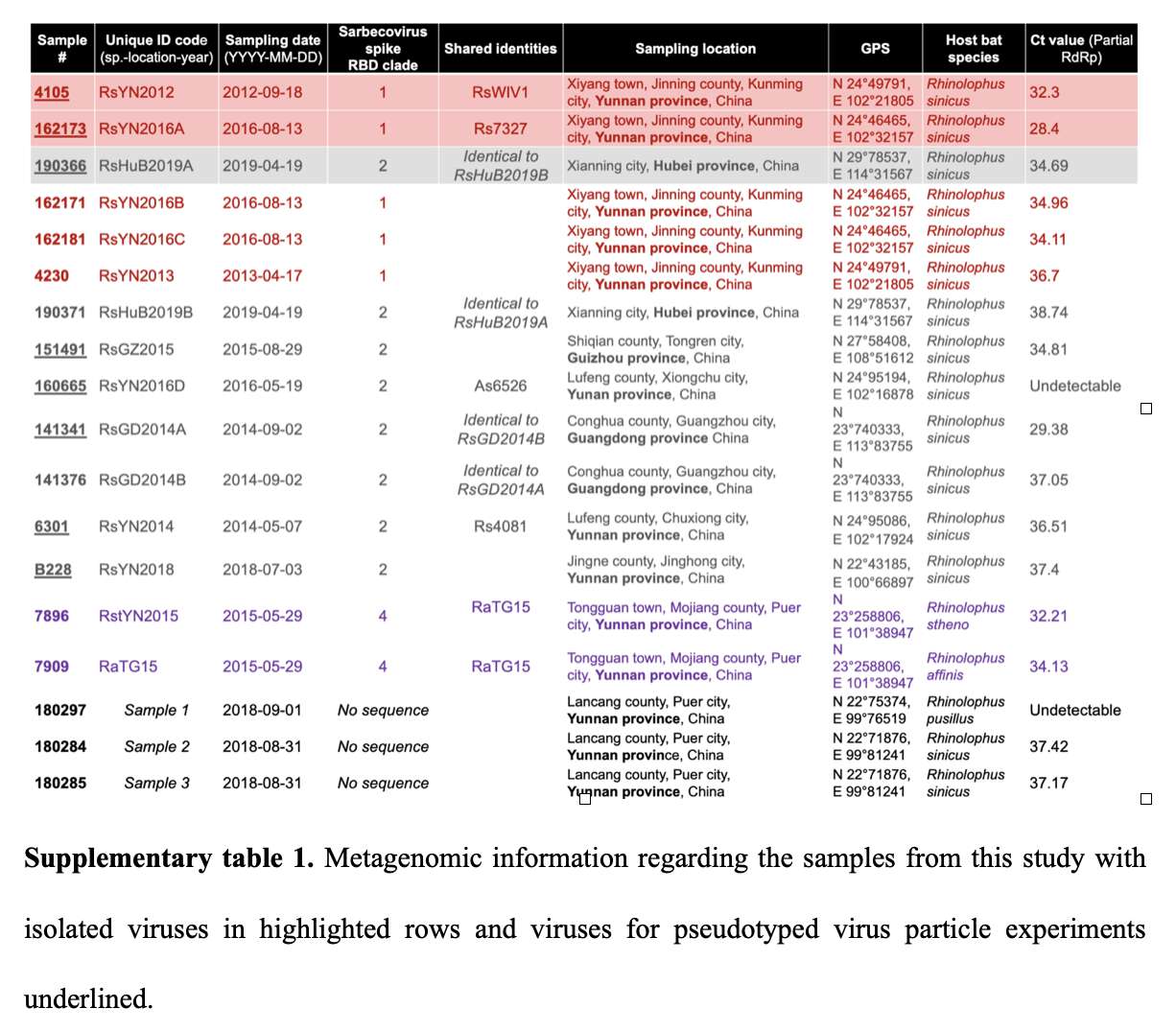
RsGD2014A seems to have some S1 recombination action going on, which is totally unexpected from a virus published by the WIV. The NTD is virtually identical to GD2017H and RaCH025:
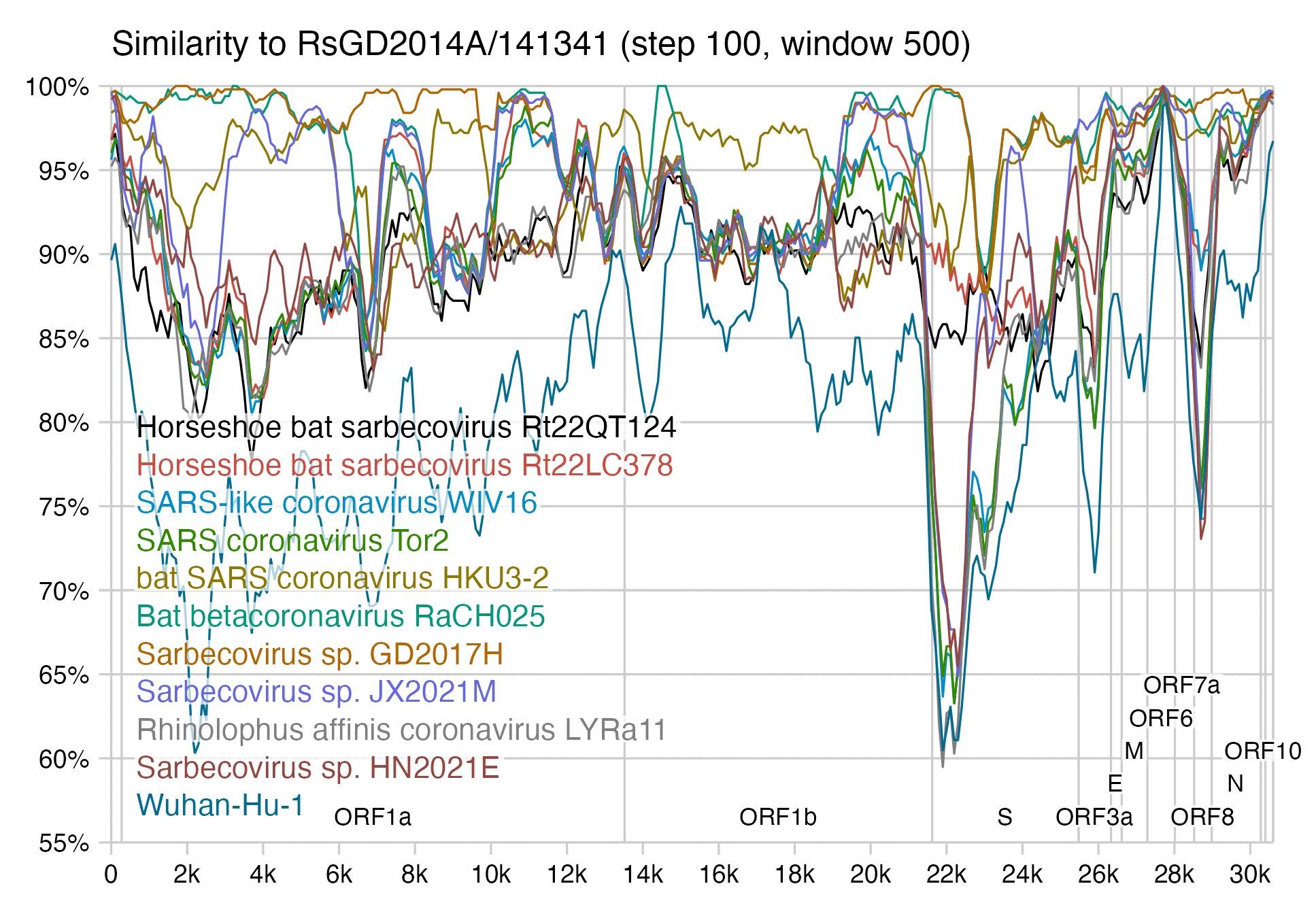
If RsGD2014A is compared to GD2017H, there are also breakpoints in the slope of cumulative synonymous mutations near the start and end of the RBD:
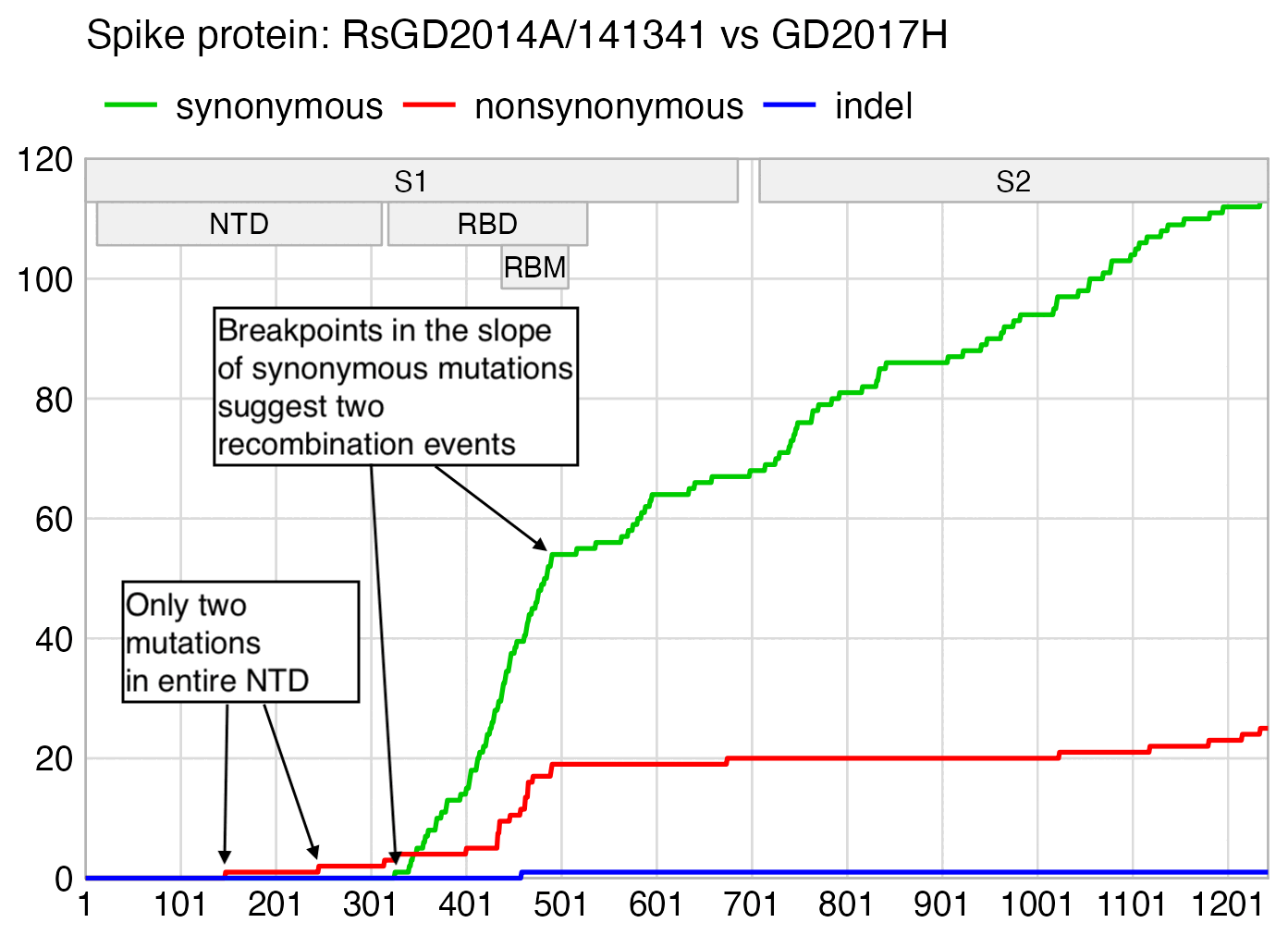
In 2021 Steven Quay et al. published a preprint which showed that in
the raw reads of samples from early COVID patients published by WIV,
there were also reads which contained the HA segment of influenza A
within a pVAC1 expression vector:
https://
The WIV reads also contained a much smaller number of reads for other
segments of H7N9, even though Quay wrote: "Mr. Cullen fails to state that the H7N9 sequences found
in the patients were one gene only, attached to a laboratory
vector." [https://
The run with the highest number of H7N9 reads has the ID WIV07-2. It has about 17,000 reads that match segment 4 but only about 150 reads which match other segments of H7N9.
The H7N9 reference sequence comes from a sample collected from a human patient in China in 2013, even though H7N9 samples from humans are otherwise fairly rare. When I aligned the reads of WIV07-2 against the refseq, I got an error rate of about 0.3% for reads that aligned against segment 4 (HA) and 0.3% for segment 8, but I got an error rate of 1.6-3.5% for the other segments (and there were only 4 reads which aligned against segment 8 so the low error rate may have been due to chance). So I thought that maybe the other segments come from a different source than segment 4:
brew install seqkit bowtie2 samtools gnu-sed curl ftp:// ftp. ncbi. nlm. nih. gov/ entrez/ entrezdirect/ install- edirect. sh| sh curl " https:// www. ebi. ac. uk/ ena/ portal/ api/ filereport? accession=SRR11092059& result=read_ run& fields=fastq_ ftp"| sed 1d| cut -f1| tr \; \\ n| sed s,^, ftp://,| xargs wget esearch -db nuccore -query '( h7n9[ organism] OR wuhan- hu- 1) AND refseq[ filter] '| efetch -format fasta| seqkit replace -isp ' a{ 5,}$ ' > h7n9sars2. fa bowtie2- build h7n9sars2. fa{,} bowtie2 -p4 -- no- unal -- local -x h7n9sars2. fa -1 SRR11092059_ 1. fastq. gz -2 SRR11092059_ 2. fastq. gz| samtools sort -@ 3 -> SRR11092059. local. bam x=SRR11092059. local. bam; samtools coverage $x| awk \$ 4| cut -f1, 3- 6|( gsed -u ' 1s/$/\ terr%\ tname/; q'; sort -rnk3| awk -F\\ t -v OFS=\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ print$ 0, sprintf( "%. 2f", a[$ 1])} ' <( samtools view $x| awk -F\\ t '{ x=$ 3; n[ x]++; len[ x]+ =length($ 10); sub(/.* NM: i:/, " "); mis[ x]+ =$ 1} END{ for( i in n) print i"\ t" 100* mis[ i]/ len[ i]} ') -| awk -F\\ t ' NR==FNR{ a[$ 1] =$ 2; next}{ print$ 0"\ t" a[$ 1]} ' <( seqkit seq -n h7n9sars2. fa| gsed ' s/ /\ t/; s/,.*// ') -)| column -ts$ '\ t'
#rname endpos numreads covbases coverage err% name NC_ 026425. 1 1708 17346 1685 98. 6534 0. 31 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 4 hemagglutinin (HA) gene NC_ 045512. 2 29870 5214 29843 99. 9096 0. 22 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan- Hu- 1 NC_ 026422. 1 2280 107 2201 96. 5351 3. 29 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 1 polymerase PB2 (PB2) gene NC_ 026427. 1 980 27 543 55. 4082 2. 57 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 7 matrix protein 2 (M2) and matrix protein 1 (M1) genes NC_ 026423. 1 2297 10 671 29. 212 3. 47 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 2 polymerase PB1 (PB1) and PB1- F2 protein (PB1- F2) genes NC_ 026428. 1 841 4 140 16. 6468 0. 33 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 8 nuclear export protein (NEP) and nonstructural protein 1 (NS1) genes NC_ 026426. 1 1499 4 349 23. 2822 2. 00 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 5 nucleocapsid protein (NP) gene NC_ 026424. 1 2176 2 207 9. 51287 1. 67 Influenza A virus (A/ Shanghai/ 02/ 2013( H7N9)) segment 3 polymerase PA (PA) and PA- X protein (PA- X) genes
Next I tried aligning the WIV07-2 reads against all H7N9 sequences
from the NCBI's influenza virus database:
https://
#rname endpos numreads covbases coverage err% name KF021597 1708 621 1685 98. 6534 0. 34 A/ Shanghai/ 02/ 2013; 4; China; 2013; 03; 05; Human CY146908 1683 603 1674 99. 4652 0. 32 A/ chicken/ Guangdong/ SD641/ 2013; 4; China; 2013; 05; 03; Avian KJ395948 1683 600 1594 94. 7118 0. 26 A/ chicken/ Guangdong/ G2/ 2013; 4; China; 2013; 05; 05; Avian KP413700 1683 578 879 52. 2282 0. 26 A/ chicken/ Dongguan/ 4195/ 2013; 4; China; 2013; 12; 19; Avian KM879322 1683 325 1505 89. 4236 0. 28 A/ chicken/ Zhejiang/ DTID- ZJU06/ 2013; 4; China; 2013; 12;; Avian
However segment 1 got most aligned reads against samples from 2017 (top 5 samples shown):
#rname endpos numreads covbases coverage err% name MG575543 2280 50 1866 81. 8421 1. 69 A/ chicken/ Anhui/ AH395/ 2017; 1; China; 2017; 03; 06; Avian KY621539 2341 8 279 11. 918 1. 08 A/ Qingyuan/ GIRD01/ 2017; 1; China; 2017; 01; 14; Human MF630106 2280 6 254 11. 1404 1. 00 A/ chicken/ Guangdong/ SD027/ 2017; 1; China; 2017; 01; 18; Avian KY643838 2340 6 484 20. 6838 1. 33 A/ Guangdong/ SP440/ 2017; 1; China; 2017; 01; 04; Human MF630458 2280 4 248 10. 8772 3. 67 A/ duck/ Hunan/ S40506/ 2015; 1; China; 2015; 11; 16; Avian
Segment 7 also seems to match newer samples from around the year 2017:
#rname endpos numreads covbases coverage err% name MW100646 1027 4 247 24. 0506 1. 50 A/ chicken/ Hunan/ 5. 24_ YYGK49P2- OC/ 2017; 7; China; 2017; 05; 24; Avian MZ803120 1000 2 227 22. 7 0. 00 A/ mallard/ South Korea/ 20X- 20/ 2021; 7; South Korea; 2021; 01; 06; Avian MW265394 982 2 160 16. 2933 1. 00 A/ chicken/ Shanxi/ SX0256/ 2019; 7; China; 2019; 01; 10; Avian MW109507 1027 2 125 12. 1714 0. 67 A/ environment/ Jiangxi/ 2. 06_ SRGFYF005- E/ 2017; 7; China; 2017; 02; 06; Environment MW109475 982 2 226 23. 0143 1. 00 A/ environment/ Jiangxi/ 1. 11_ NCDZT98F2- E/ 2017; 7; China; 2017; 01; 11; Environment
So it could be that the HA plasmid was designed based on the refseq strain from 2013, but the full virus came from a newer strain of H7N9.
Steve Massey has been saying that SARS 2 is phylogenetically closer
to RaTG13 than to BANAL-52 because it groups together with RaTG13 in
maximum likelihood trees: [https://

However the same thing can even happen in a distance-based tree. The tree below is based on a percentage distance of the whole genome so that positions where either sequence has a gap are not considered. I used the complete linkage method where the distance between two branches is the distance between the furthest nodes within the branches, so that for example the distance between SARS2 and the branch that contains the three BANAL viruses is the distance between SARS2 and the BANAL virus which is the furthest from SARS2, which happens to be BANAL-236. In my tree first BANAL-103 is joined with BANAL-236, and then their combined branch is joined with BANAL-52. And after that Wuhan-Hu-1 ends up being closer to RaTG13 than to the branch of the three BANAL viruses, so Wuhan-Hu-1 joins RaTG13 earlier:
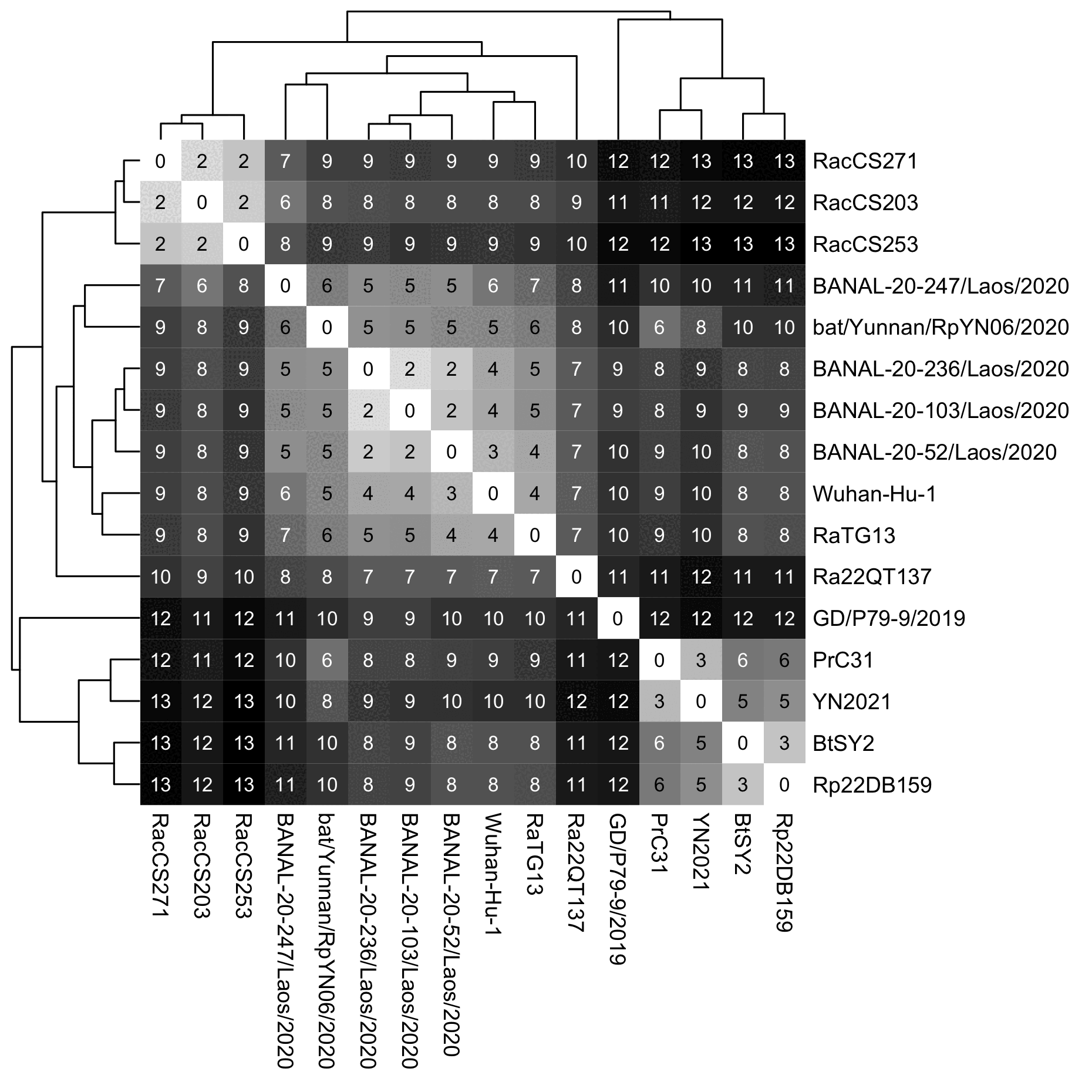
The branching pattern remained the same even when I changed the
linkage method from complete to ward.. But
when I removed BANAL-236 and BANAL-103 from the tree, then Wuhan-Hu-1
joined with BANAL-52 before RaTG13:
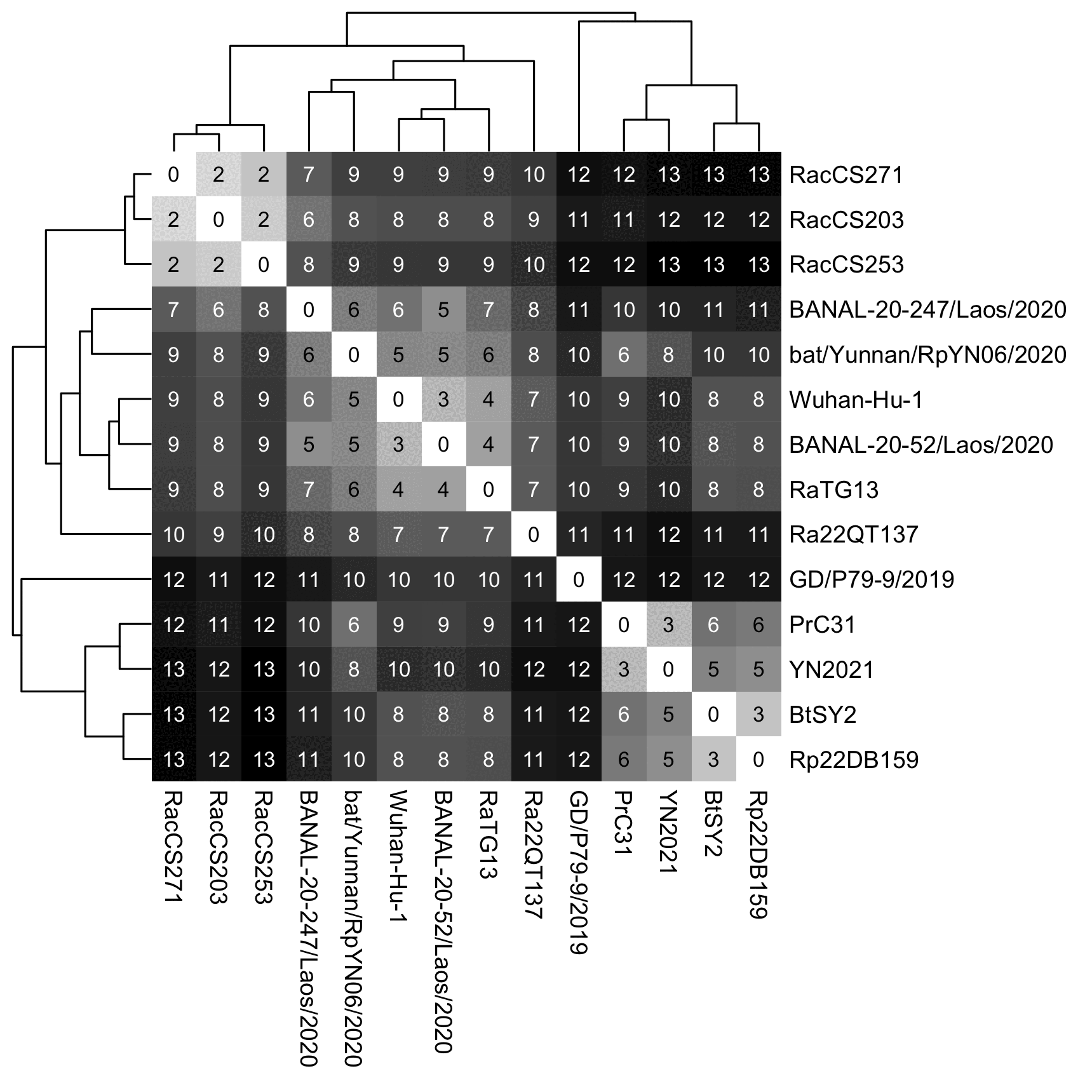
brew install iqtree mafft seqkit xz wget sars2.net/ f/ sarbe.{ pid, fa. xz}; xz -d sarbe. fa. xz # thin out a TSV percentage identity matrix to remove sequences with over n% thin()(identity to any previous sequence (default 99%) awk -F\\ t ' NR> 1{ for( i=2; i< NR; i++) if($ i> x) next; print$ 1} ' x="${ 1- 99} " "${@: 2} ") # matrix sort grep (sort TSV matrix by column that matches regex and print the column along with the first column) masog()(awk -F\\ t ' NR==1{ for( i=2; i< =NF; i++) if($ i~ x) break; next}{ print$ i,$ 1} ' x="$ 1" "${ 2--} "| sort -n) # short name shorn()(sed -E $' s/(,| surface| ORF1| orf1| genomic sequence).*//; s/ Severe acute respiratory syndrome/ SARS/; s/ isolate / /') shorn2()( shorn| sed ' s/ RNA$//; s/ >.* />/ ') # select sequences with at least 92% masog Hu-identity to Wuhan- Hu- 1; omit sequences with over 99. 9% identity to another sequence 1< sarbe. pid| awk '$ 1> =90{ print$ 2} '| sort| comm -12 <( thin 99. 9 sarbe. pid| cut -d\ -f1| sort) -| seqkit grep -f- sarbe. fa| shorn2| mafft -- thread 4 -- quiet -> iqtree. fa seqkit grep -nvrp ' 20-( 103| 236) ' iqtree. fa> iqtree2. fa curl -Ls sars2. net/ f/ pid. cpp> pid. cpp; g++ pid. cpp -O3 -o pid pid< iqtree. fa> iqtree. pid; pid< iqtree2. fa> iqtree2. pid
t=100-read. table( " iqtree. pid", row. names=1, header=T, sep="\ t", check. names=F) m=as. matrix( t) hc=hclust( as. dist( m)) # complete linkage #hc=hclust( hc=as.as. dist( t), " ward. D2") # Ward linkage hclust( reorder( as. dendrogram( hc), cmdscale( m)[, 1])) pheatmap:: pheatmap( m, filename=" 1. png", display_ numbers=round( m), legend=F, clustering_ callback=\(...) hc, cellwidth=17, cellheight=17, fontsize=9, fontsize_ number=8, border_ color=NA, number_ color=ifelse( t> max( t)*. 45&! is. na( t), " white", " black")[ hc$ order, hc$ order], sapply( 255: 0,\( i) rgb( i, i, i, maxColorValue=255)))
In a world where more RaTG13-like viruses would be published, RaTG13 might join other RaTG13-like viruses earlier than SARS2. So then if the other RaTG13-like viruses would be further from SARS2 than RaTG13 is, it would increase the distance between SARS2 and the branch of RaTG13-like viruses, so then SARS2 might join the BANAL branch before the branch of RaTG13-like viruses. So therefore which branch is joined by SARS2 first might depend on which branch has more published sequences.
In the output below I used IQTree to generate maximum likelihood trees. On the left side where I included all 5 BANAL viruses, Wuhan-Hu-1 is grouped together with RaTG13. But on the right side where I again removed BANAL-103 and BANAL-236, Wuhan-Hu-1 is grouped together with BANAL-52 and not RaTG13. So a similar phenomenon I demonstrated above using a distance-based tree can also occur in a maximum likelihood tree:
$iqtree -nt AUTO -s iqtree. fa; iqtree -nt AUTO -s iqtree2. fa [...] $ paste <( echo $'--- All five BANAL viruses included ---\ n'; sed ' 1,/^ NOTE:/ d' iqtree. fa. iqtree| sed 1d| sed '/^$/ q') <( echo $'--- BANAL- 103 and BANAL- 236 excluded ---\ n'; sed ' 1,/^ NOTE:/ d' iqtree2. fa. iqtree| sed 1d| sed '/^$/ q')| column -ts$ '\ t' --- All five BANAL viruses included --- --- BANAL- 103 and BANAL- 236 excluded --- +** Ra22QT137 +** Ra22QT137 | | +** Ra22QT106 +** Ra22QT106 | | | +-- Ra22QT135 | +-- Ra22QT135 +**| +**| | +-- Ra22QT77 | +-- Ra22QT77 +--| +--| | +-- Wuhan- Hu- 1 | +-- Wuhan- Hu- 1 | +--| | +--| | | +----- RaTG13 | | +-- BANAL- 20- 52_ Laos_ 2020 | +--| | +--| | | +-- BANAL- 20- 52_ Laos_ 2020 | | +----- RaTG13 | +--| | +--| | | | +-- BANAL- 20- 236_ Laos_ 2020 | | | +-- bat_ Yunnan_ RpYN06_ 2020 | | +--| | | | +---| | | +-- BANAL- 20- 103_ Laos_ 2020 | | | | | +-- PrC31 | +--| | | | | +---------------| | | | +-- bat_ Yunnan_ RpYN06_ 2020 | | | | | +-- YN2021 | | | +---| | | | | +---| | | | | | +-- PrC31 | | | | | +-- Rp22DB159 | | | | +--------------| | | | | +--------| | | | | | +-- YN2021 | | | | +-- BtSY2 | | | | +---| | | +--| | | | | | +-- Rp22DB159 | | | +-- BANAL- 20- 247_ Laos_ 2020 | | | | +--------| | | | +---| | | | | +-- BtSY2 | | | | +-- BANAL- 20- 116_ Laos_ 2020 | | +--| | | +----| | | | +-- BANAL- 20- 247_ Laos_ 2020 | | | +-- RacCS271 | | | +--| | | +------------| | | | | +-- BANAL- 20- 116_ Laos_ 2020 | | | +-- RacCS253 | | +----| | | +--| | | | +-- RacCS271 | | +-- RacCS203 | | +------------| +--------------| | | | +-- RacCS253 | +-- GD_ P79- 9_ 2019 | | +--| +-------------------------| | | +-- RacCS203 +-- MP789 +---------------| | +-- GD_ P79- 9_ 2019 +-----------------------| +-- MP789
LongDesertTrain posted this tweet: [https://

In a nearly complete collection of GISAID submissions with a collection date in 2020, there were a total of 10,005 unique insertions. However G was the least common nucleotide within in the insertions, even though C is less common than G in Wuhan-Hu-1:
$curl -Ls sars2. net/ f/ gisaid2020. tsv. xz| xz -dc> gisaid2020. tsv $ cut -f14 gisaid2020. tsv| tr , \\ n| awk '! a[$ 0]++ '| cut -d: -f2| grep -o '[ ACGT] '| sort| uniq -c| sort 26495 G 28057 C 39400 T 43419 A $ curl ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=fasta& id=MN908947' > sars2. fa $ seqkit seq -s sars2. fa| grep -o .| sort| uniq -c| sort 5492 C 5863 G 8954 A 9594 T
Even when I only looked at insertions which occurred in at least 10 submissions, G was still the least common:
$cut -f14 gisaid2020. tsv| sed 1d| tr , \\ n| awk '++ a[$ 0] ==10'| grep -o '[ ACGT] '| sort| uniq -c| sort 283 G 304 C 505 T 544 A
Steve Massey posted a Twitter thread where he suggested that the
RaTG13 sample may have come from a vaginal plug of a female bat that
contained stored sperm. [https://
You can try out the utility if you copy the names of the thousand
genes with the highest z-score for RaTG13 from here:
https://
However RaTG13 is listed as a sample from a male bat at GISAID. [https://
In order to guess the sex of the sample, I tried aligning all reads
of RaTG13 against a Y-chromosome sequence of Rhinolophus
affinis:
https://
I downloaded the first million reads of RaTG13, the RaTG15-related
runs from CNCB, the WIV bat anal swab runs from February 2020, and a
couple of other R. affinis runs. [https://
In a sample from a female bat that had some sperm cells mixed in, the percentage of Y-chromosome reads might be expected to be intermediate between a sample from a male bat and a sample from a female bat. However RaTG13 ended up getting one of the highest percentages of Y-chromosome reads, which I think makes it less likely that the sample was actually from a female bat. Here the second column shows the number of reads that aligned against the y-chromosome, and the third column shows the number of reads that aligned against all chromosomes, and the first chromosome shows the ratio between the second and third columns as a percentage:
$datasets download genome accession GCA_ 040057535. 1 -- include genome [...] $ unzip ncbi_ dataset. zip [...] $ seqkit grep -nrvp contig ncbi_ dataset/*/*/*. fna> raffinis. fa $ mkdir ratg15; parallel -j10 curl -Ls https:// download. cncb. ac. cn/ gsa/ CRA004339/{}/{}_ f1. fq. gz\| seqkit head -n1000000 -o ratg15/{}. fastq. gz ::: CRR29060{ 0.. 7} $ printf %s\\ n SRR110857{ 97, 33.. 41} SRR1206284{ 4, 5} SRR22936{ 563, 575, 577, 578, 715, 806}| parallel -j20 fastq- dump -O wivswab -X 1000000 -- gzip {} [...] $ for f in {wivswab, ratg15}/* gz; do minimap2 -- secondary=no -a -- sam- hit- only raffinis. fa $f| samtools view -b ->${ f% fastq. gz} bam; done [...] $ for f in {wivswab, ratg15}/* bam; do samtools view -F2048 -q40 $f| awk '$ 3==" CM078718. 1"{ y++} END{ print y/ NR* 100, y, NR, x} ' x=${ f##*/}; done| sort -n| column -t 0. 0000 0 10021 SRR11085735 # 2020 Hipposideros pomona 0.0000 0 10985 SRR11085740 # 2020 Miniopterus pusillus 0.0000 0 117541 SRR11085738 # 2020 Pipistrellus abramus 0.0000 0 126057 SRR11085739 # 2020 Tylonycteris pachypus 0.0000 0 127436 SRR11085734 # 2020 Miniopterus schreibersii 0.0000 0 259098 SRR11085733 # 2020 Hipposideros larvatus 0.0000 0 28214 SRR11085741 # 2020 Rousettus aegyptiacus 0.0000 0 77898 SRR11085736 # 2020 Rhinolophus affinis 0.0000 0 92441 SRR11085737 # 2020 Scotophilus kuhlii 0.0014 2 147779 SRR22936563 # 2023 R. 0.affinis rectum 0040 30 745986 CRR290604 # CNCB Rs7921 0.0063 54 857367 CRR290602 # CNCB Rs7907 0.0121 104 856451 SRR22936806 # 2023 R. 0.affinis rectum 0182 151 831478 SRR22936577 # 2023 R. 0.affinis rectum 0288 250 868272 SRR22936578 # 2023 R. 0.affinis rectum 0327 248 758136 SRR12062845 # male R. 0.affinis 0364 290 797144 SRR12062844 # male R. 0.affinis 0685 606 884761 SRR22936575 # 2023 R. 0.affinis rectum 0808 655 810586 SRR22936715 # 2023 R. 0.affinis rectum 0959 858 895039 CRR290603 # CNCB RaTG15 0.2698 2258 836945 CRR290600 # CNCB Rs7896 0.3205 957 298629 SRR11085797 # RaTG13 0.3283 2712 826136 CRR290605 # CNCB Rs7924 0.3584 2908 811415 CRR290606 # CNCB Rs7931 0.3661 2984 815123 CRR290601 # CNCB Rs7905 0.3990 3275 820898 CRR290607 # CNCB Rs7952
In the code above the -- flag disables
secondary alignments, but it doesn't disable supplementary alignments
which get later filtered out by -.
A binary for the datasets command can be downloaded from
here:
https://
In 2024 Alexandre Hassanin and coauthors published a paper where they
described a new SADS-related bat virus from Vietnam, and they compared
it to earlier Chinese SADS-related bat viruses and SADS. [https://
They pointed out that several of the Chinese viruses displayed unnatural recombination patterns which may have been inauthentic (even though the authors seem to have played it safe by suggesting it was the result of unintentional mistakes and not deliberate fraud):
By examining SWB bipartitions showing strong support in these two regions, we found that two Rhinacovirus genomes sequenced from R. pusillus are problematic, RpGX16-Q222 and RpGX17-Q225 (GenBank accessions OQ175208 and OQ175209, respectively [9]): they appeared to be closely related to all SADS-CoVs in positions 14,601-14,900 (SWB400 bipartition n°836) and to all SADS-CoVs except CH/FJWT/2018 in positions 21,501-21,900 (SWB400 bipartition n°571) (Figure S6); by examining the alignment, we found that they are identical to most SADS-CoVs in positions 14,503-15,555, whereas in positions 21,265-21,959, they are identical to SADS-CoV CHN-GD/2017 and very similar to other SADS-CoVs (99.7% of identity) (Figure 6A). By contrast, both RpGX16-Q222 and RpGX17-Q225 appeared to be very divergent from other members of the R. pusillus group in these two regions: between 14% and 15% in the first region and between 27% and 29% in the second region. If we consider RpGX16-Q222 and RpGX17-Q225 genomes to be entirely authentic, we have to admit that a SADS-CoV strain may have recombined with a bat rhinacovirus of the R. pusillus group. Such a scenario seems very unlikely as it implies that either pigs or R. pusillus bats were coinfected by SADS-CoV and a rhinacovirus of the R. pusillus group. As an alternative, we rather suggest that the genomes of RpGX16-Q222 and RpGX17-Q225 are chimeric: although most parts of their genomes are authentic sequences, two regions were mistakenly assembled with SADS-CoV sequences (Figure 6A).
[...]
Our analyses revealed that RaYN20-Q207 (GenBank accession: OQ175197 [9]) is another problematic genome. Whereas most parts of its genome were found to support a grouping with RaYN20-Q216 (GRPS = 40%; Figure 3), three fragments of ORF1ab and one fragment of the Spike gene rather provided support for a grouping with RaGD17-Q208 (GRPS = 28%; Figure 5). By examining the alignment, we found a perfect identity between RaYN20-Q207 and RaGD17-Q208 in three genomic regions, i.e., positions 1-1787 (length: 1787 nt), 3820-7943 (length: 4124 nt), and 19,806-20,198 (length: 393 nt), whereas adjacent genomic regions showed much more nucleotide divergence (Figure S7). If these two sequences are authentic, our interpretation is that these three fragments were inherited in the ancestor of RaYN20-Q207 by genomic recombination with a parental virus identical or closely related to RaGD17-Q208. However, this hypothesis seems unlikely, as RaGD17-Q208 was sampled in 2017 in Guangdong, whereas RaYN20-Q207 was collected in 2020 in Yunnan. As an alternative hypothesis, we propose that RaYN20-Q207 is a chimeric genome containing three misassembled fragments from RaGD17-Q208.
[...]
The 5'-end of CHN-GD/2017 (positions 1-1787) was found to be identical to that of RsHKU2 (GenBank accession: NC_009988) (Figure 6A), whereas these two viruses were collected from a neonatal piglet with acute diarrhoea in 2017 and from a R. sinicus in 2006, respectively [12,27]. Since 11 years of evolution without any mutations seems inconceivable, we conclude that the 5'-end of CHN-GD/2017 is not authentic as it was completed with that of the RsHKU2 genome, which was sequenced and published several years before.
[...]
Although these results may suggest that Ra162140 was a direct parental progenitor of SADS-CoV CH/FJWT/2018, we consider this hypothesis to be unlikely since Ra162140 was collected between 2013 and 2016 from an R. affinis of Guangdong Province, China [3,28], whereas SADS-CoV CH/FJWT/2018 was collected in 2018 from a diarrhoeal piglet of Fujian Province, China [5]. After at least two years of evolution and a host species jump from bats to pigs, we indeed expect to find a few mutations separating SADS-CoV CH/FJWT/2018 from Ra162140 in the two problematic regions of 1890 and 703 nt, respectively. As an alternative hypothesis, we, therefore, suggest that SADS-CoV CH/FJWT/2018 could be a chimeric genome: although most parts represent an authentic SADS-CoV sequence, two regions were potentially completed with Ra162140 sequences.
[...]
According to the hypothesis involving recombination, CH/FJWT/2018 and Guangxi/2021 genomes are considered to be fully authentic; based on the three genomic fragments A, B and B' shown in Figure S6, we can propose that at least three recombination events occurred in pigs and involved the main SADS-CoV lineage (the one containing all strains currently sequenced except CH/FJWT/2018 and Guangxi/2021) and a second swine lineage identical or more closely related to the bat rhinacovirus Ra162140. According to the hypothesis involving human errors, CH/FJWT/2018 and Guangxi/2021 genomes are considered to be artificially chimeric; the fragment(s) of bat rhinacoviruses found in their genomes were not authentic; they were integrated into the sequences following human errors, which could be due to carry-over contamination during PCRs, incorrect pooling of pig and bat samples in the same NGS library, or bioinformatic error (e.g., the consensus sequence was misassembled by calling a bat reference sequence when there was no coverage). To provide definitive conclusions on the authenticity of their genomes, we consider that [5,7] should deposit their metagenomic data and Sanger electropherograms in international databases and publish the primer sets they used for PCR amplification and sequencing. In the absence of these data, these two problematic SADS-CoV genomes should be considered doubtful and excluded from future studies.
Hassanin et al. also wrote:
Recently, [33] reported that SADS-CoV re-emerged in June 2023 in Henan Province (central China) with an outbreak that caused the death of about 400 piglets. The genome of the new SADS-CoV strain (HNNY/2023; GenBank accession: PP069800 [33]) shared 99.92% of nucleotide identity with the first SADS-CoVs identified in Guangdong in 2017 [3]. Such a high nucleotide conservation means that all SADS-CoV strains originated from the same source, i.e., farmed pigs contaminated by bats in northern Guangdong, and that the virus has been circulating silently in domestic pigs for at least seven years, spreading from farm to farm in Guangdong between 2016 and 2019 [1,2,3,4], and then in the west in Guangxi in 2021 [7], and in the north in Jiangxi and Fujian in 2018 [5,10], and more recently in Henan in 2023 [33].
The new SADS-CoV sequence HNNY/2023 has an assembly error at the 5' end where bases 1-54 are identical to bases 3193-3246:
$curl ' https:// eutils. ncbi. nlm. nih. gov/ entrez/ eutils/ efetch. fcgi? db=nuccore& rettype=fasta& id=PP069800' > newsads. fa $ seqkit subseq -r1: 54 newsads. fa| seqkit locate -f- newsads. fa |cut -f1, 4- 6| column -t seqID strand start end PP069800. 1 + 1 54 PP069800. 1 + 3193 3246
When I excluded the poly(A) tail and the region with the assembly error in the 5' UTR, HNNY/2023 had 10 bases different from SADS-CoV/CN/GDWT/2017, 9 bases different from SADS-CoV/GDWT-P7 (7th passage), and 8 bases different from SADS-CoV/GDWT-P18 (18th passage, with a collection date in March 2018):
#top 250 BLAST matches in 2025 to new SADS sequence from 2023 curl -Ls sars2.net/ f/ sads25. fa. xz| xz -dc> sads25. fa # nucleotide changes from first sequence in FASTA file to each other sequence dist1u()(seqkit fx2tab "$@ "| awk -F\\ t ' NR==1{ split($ 2, a, " "); l=length( a); next}{ split($ 2, b, " "); d=0; for( i=1; i< =l; i++) if( a[ i]! =b[ i]) d++; print d,$ 1} ') # closest neighbors of new 2023 SADS sequence with ends of UTRs excluded seqkit subseq -r 59:34670 sads25. fa| seqkit fx2tab| sed 1p| seqkit tab2fx| dist1u| sort -n| head -n16| cut -d\| -f- 3| sed ' s/ Swine acute diarrhea syndrome coronavirus/ SADS- CoV/; s/ isolate / /'| sed ' s/ /|/; s/ /|/ '|( echo ' muts| accession| name| creation| collection'; cat)| tr \| \\ t| csvtk -t pretty -s\ |sed 2d
muts accession name creation collection 0 PP069800.1 SADS- CoV strain SADS- CoV/ HNNY/ 2023 2024- 03- 27 2023- 06- 20 8 MK994935. 1 SADS- CoV SADS- CoV/ GDWT- P18 2020- 03- 03 2018- 03 # passage 18 of cultured strain 9 MK994934.1 SADS- CoV SADS- CoV/ GDWT- P7 2020- 03- 03 2017 # passage 7 of cultured strain 10 MG557844.1 SADS- CoV SADS- CoV/ CN/ GDWT/ 2017 2018- 03- 18 2017- 04 # original pig sample from 2017 10 MT039231.1 Mutant SADS- CoV strain icSADS 2020- 06- 15 12 MF769420. 1 SADS- CoV 246 2018- 03- 18 2017 13 MF769424. 1 SADS- CoV 182 2018- 03- 18 2017 14 MF094683. 1 SADS- CoV FarmC 2018- 03- 18 14 MW727454. 1 Porcine enteric alphacoronavirus GDZQ- 2018 2022- 03- 01 2018- 09- 01 15 MF094682. 1 SADS- CoV FarmB 2018- 03- 18 15 MF769417. 1 SADS- CoV 31 2018- 03- 18 2017 15 MG605091. 1 SADS- CoV SADS- CoV/ CN/ GDLS/ 2017 2018- 09- 16 2016- 08- 09 16 MF094681. 1 SADS- CoV FarmA 2018- 03- 18 16 MF769428. 1 SADS- CoV 53 2018- 03- 18 2017 17 MF769419. 1 SADS- CoV 197 2018- 03- 18 2017 17 MF769441. 1 SADS- CoV 82 2018- 03- 18 2017
So was there convergent evolution in cell culture and pig farms, where the cultured strain gradually evolved closer to the strain that was detected on pig farms in 2023? There should normally be much more than 8-10 mutations over a period of about 5-6 years.
The SADS-CoV/HNNY/2023 sequence was described in a paper titled "Re-emergence of severe acute diarrhea syndrome
coronavirus (SADS-CoV) in Henan, central
China, 2023". The paper is behind a paywall, but the parts of the
paper that are freely visible seem to have suggested the virus developed
naturally out of a virus that had been circulating on pig farms since
the 2017 outbreak: [https://
Severe acute diarrhea syndrome coronavirus (SADS-CoV) was first detected in Guangdong province of China in 2017. And yet from May 2021 to Jun 2023, there were no SADS-CoV outbreaks. In this study, we reported the recent outbreak of SADS-CoV in China on Jun 2023. Phylogenetic analysis showed the novel strain was derived from the ongoing transmission and evolution of SADS-CoV in China, rather than a separate cross-species transmission from bats. Also, the novel strain was found to participate in a recombant event as a minor parent and a missing base in the genome was discovered indicating an novel evolutionary pathway.
The "missing base" probably refers to a single gap at position 26,714 in my alignment here, which results in a premature termination of the last ORF. This shows the new sample from 2023 on the left and the 18th passage of the 2017 sample on the right:
$R -e ' if(! require( " BiocManager")) install. packages( " BiocManager"); BiocManager:: install( " Biostrings") ' $ aldi2(){ cat "$@ " >/ tmp/ aldi; Rscript -- no- init- file -e ' m=t( as. matrix( Biostrings:: readAAStringSet( "/ tmp/ aldi"))); diff=which( rowSums( m! =m[, 1]) > 0); split( diff, cumsum( diff( c(- Inf, diff))! =1))| > sapply(\( x) c( if( length( x) ==1) x else paste0( x[ 1], "- ", tail( x, 1)), apply( m[ x,, drop=F], 2, paste, collapse=" ")))| > t()| > write. table( quote=F, sep="; ", row. names=F) ';} $ seqkit grep -nrp HNNY/ 2023, GDWT- P18 sads25. fa| mafft -- quiet -- thread 4 -| cut -d\| -f- 3| aldi2 ; PP069800. 1 Swine acute diarrhea syndrome coronavirus strain SADS- CoV/ HNNY/ 2023| 2024- 03- 27| 2023- 06- 20; MK994935. 1 Swine acute diarrhea syndrome coronavirus isolate SADS- CoV/ GDWT- P18| 2020- 03- 03| 2018- 03 1- 3;-- g; aca 5; t; g 7; a; g 9- 55; a t c a t t g a t a c t g c t c t t a c t g c a a c t a a g t c a t a c t a c a a t g t t c c ;----------------------------------------------- 6649; c; t 10784; a; c 20879; a; g 21395; a; g 23506; c; a 24712; c; t 26714;-; t 26997; t; c 27213- 27263; a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a ;---------------------------------------------------