See these pages:
Fleming wrote: "Yes, @kalashnikity I received my PhD two days after my HS diploma. That was the requirement from the work begun in 1968 when our group, following the MK Ultra group, was initiated. I owe much to many including respect and confidentiality of our work." [https://x.com/Doctor_I_am_The/status/1876680552952004630, https://archive.is/IZdz7]
When someone asked Fleming to post his PhD thesis to show it wasn't fake, Fleming replied simply "Q-clearance" (which I presume meant he couldn't publish his thesis because it contained classified information). [https://x.com/Doctor_I_am_The/status/1876722383400083499] Fleming also told me: "As far as my research for the thesis, this was classified as Q-clearance in 1974. I have reached out to Trump and others to work with him using this research." [https://x.com/Doctor_I_am_The/status/1882153774636028052] And later Fleming said: "I myself agreed to make my doctorate research Q-clearance because I could not envision humanity using it for the peaceful purposes I had intended it to be used for. I could only envision it being used for destructive purposes." [https://x.com/Doctor_I_am_The/status/1893677446081269778]
The Twitter user LogarithmicDis found many problems with Fleming's diploma: [https://x.com/LogarithmicDis/status/1882133468500660348]
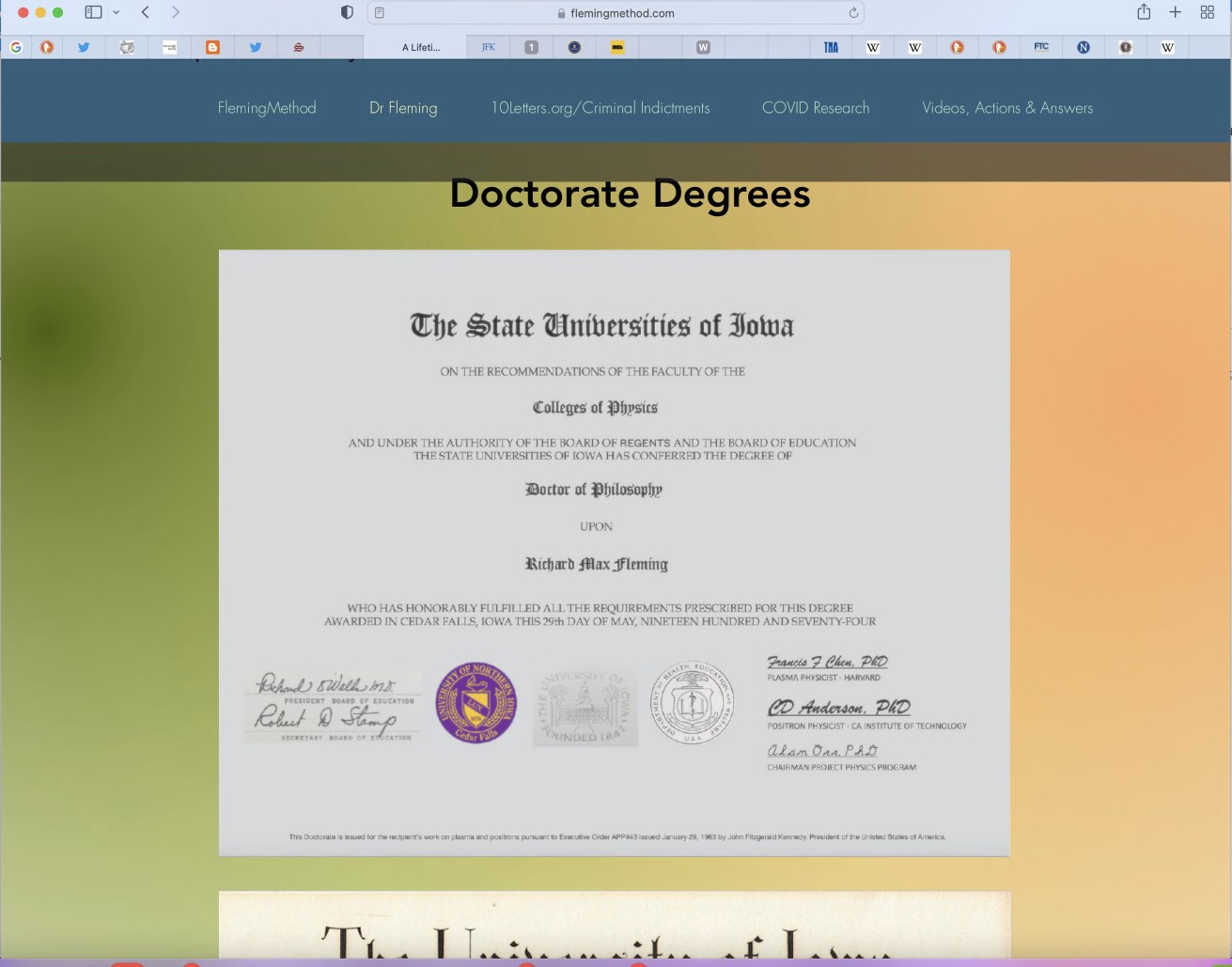
On his website he has posted his "highly unusual" PhD diploma that he started on supposedly at age 12, if you subtract his birth year (posted online by him) from a 1968 matriculating date = 12 years old.
But the diploma has MANY problems.
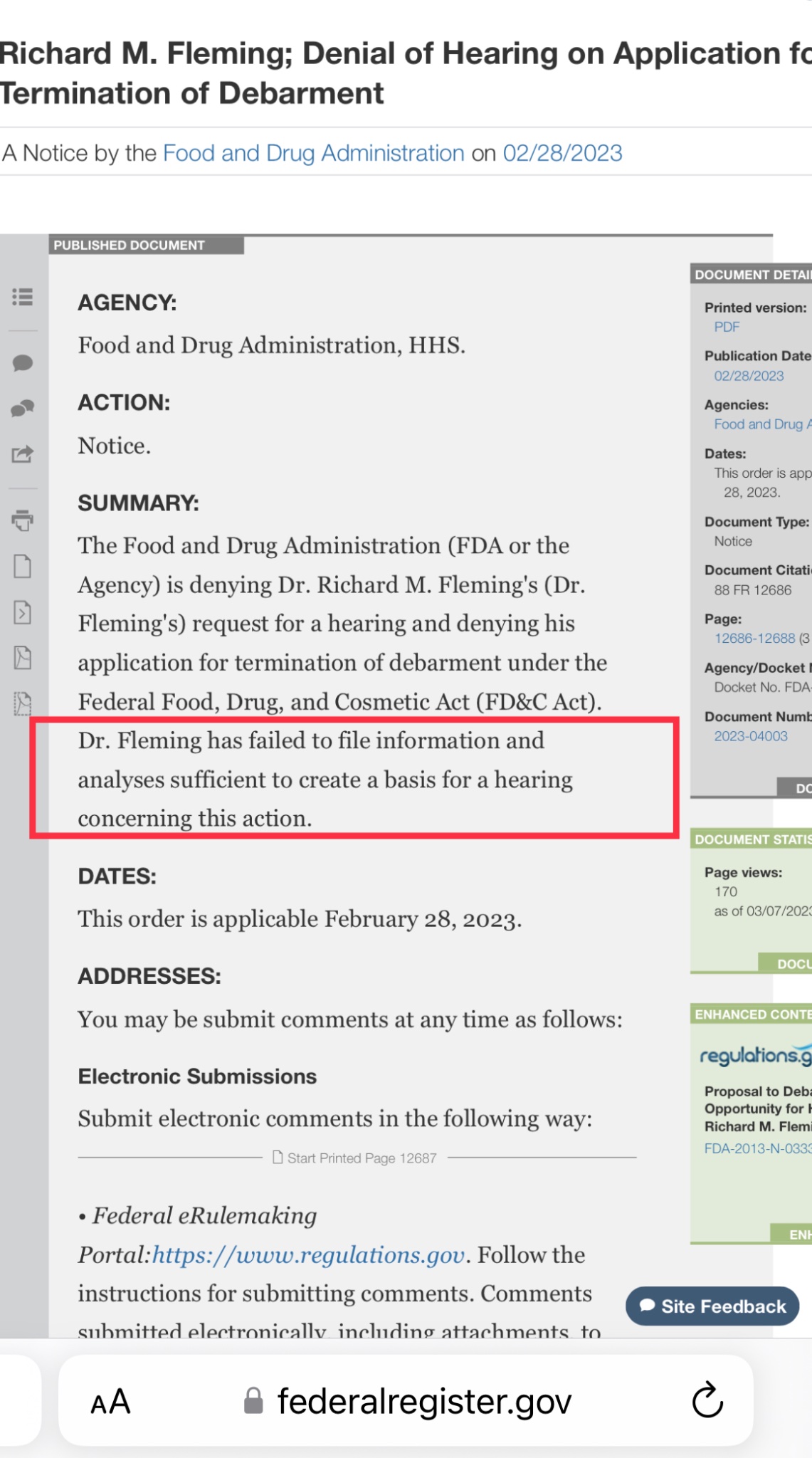
First, the doctor is a convicted felon and was sentenced for his criminal acts and debarred by the FDA.
https://archives.fbi.gov/archives/omaha/press-releases/2009/om082009.htm
So his honesty isn't exactly batting a thousand here. Upon closer look at his CV posted on his website:

And closer look at the unusual PhD diploma with contrast adjusted, we see a copy and paste left hand signature rectangle and very unusual seals.
But there are 12 total problems with this "diploma" that I see:

1. There is no such thing as "The State Universities of Iowa".
University of Iowa was in fact called "State University of Iowa".
Singular, not plural.
https://iowaregents.edu/the-board/regents-history

2. The copy & paste left hand signatures (contrast adj).
3. The president of the Board of Regents was Mary Peterson in 1974. So wrong name here.
4. The proper title is President, Board of Regents (not Board of Education, a term that ceased in 1955).
https://iowaregents.edu/the-board/regents-history

5. The seals appear to not be original to the background
6. LT and RT seals do not fade w/the document (along w/the square patch on left and e-signatures in right, addressed later).
7. The middle seal appears curved and uneven, like a poor patch job.
8. Right seal is what?
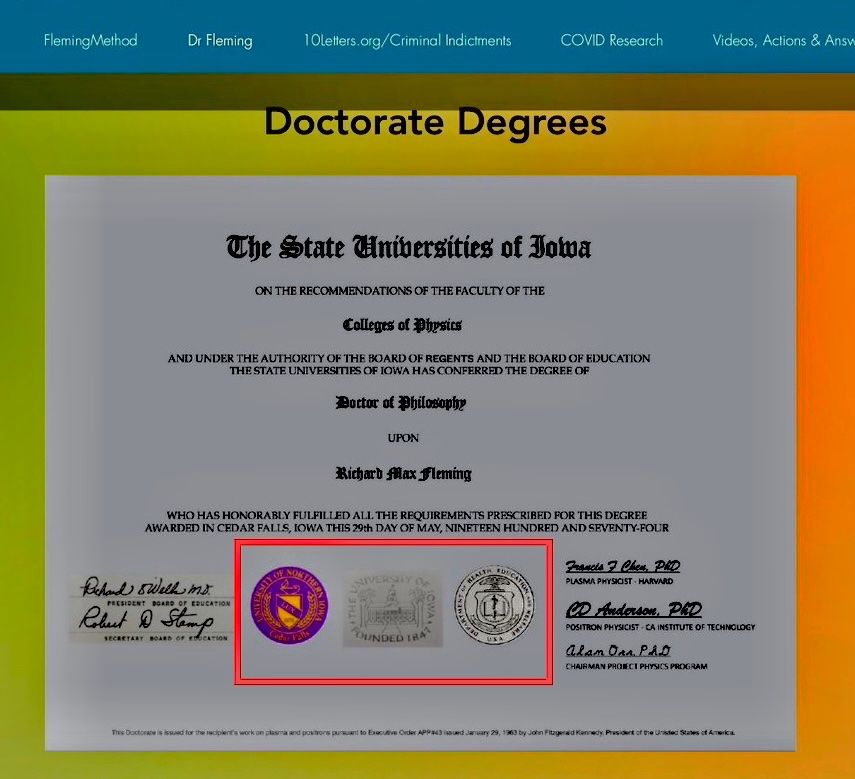
9. Two electronic signatures are exactly the same font and uncharacteristic of 1974.
10. Weird fine print at bottom with misspelling.
11. Executive order reference to JFK is strange on a diploma. Here are JFK's EOs:
https://en.wikipedia.org/wiki/List_of_executive_actions_by_John_F%2e_Kennedy

12. UNI does not confer PhD degrees. It was formerly a teachers college and confers only EdD degrees.

Here's his CV from @Doctor_I_am_The federal criminal court case in 2009.
He failed to update it.
Certification Board in Nuclear Cardiology reveals no certifications currently.
https://apca.org/certifications-examinations/cbnc-and-cbcct/cbnc/
ASNC says expired in 2002.

I verified the diploma was fake with the regent universities. I actually called (like that was necessary? lol 😂)
[...]
And the ridiculous statement at the bottom of Fleming's confirmed fake degree is a complete joke.
Here are JFK's EO's that day.
https://federalregister.gov/presidential-documents/executive-orders/john-f-kennedy/1963
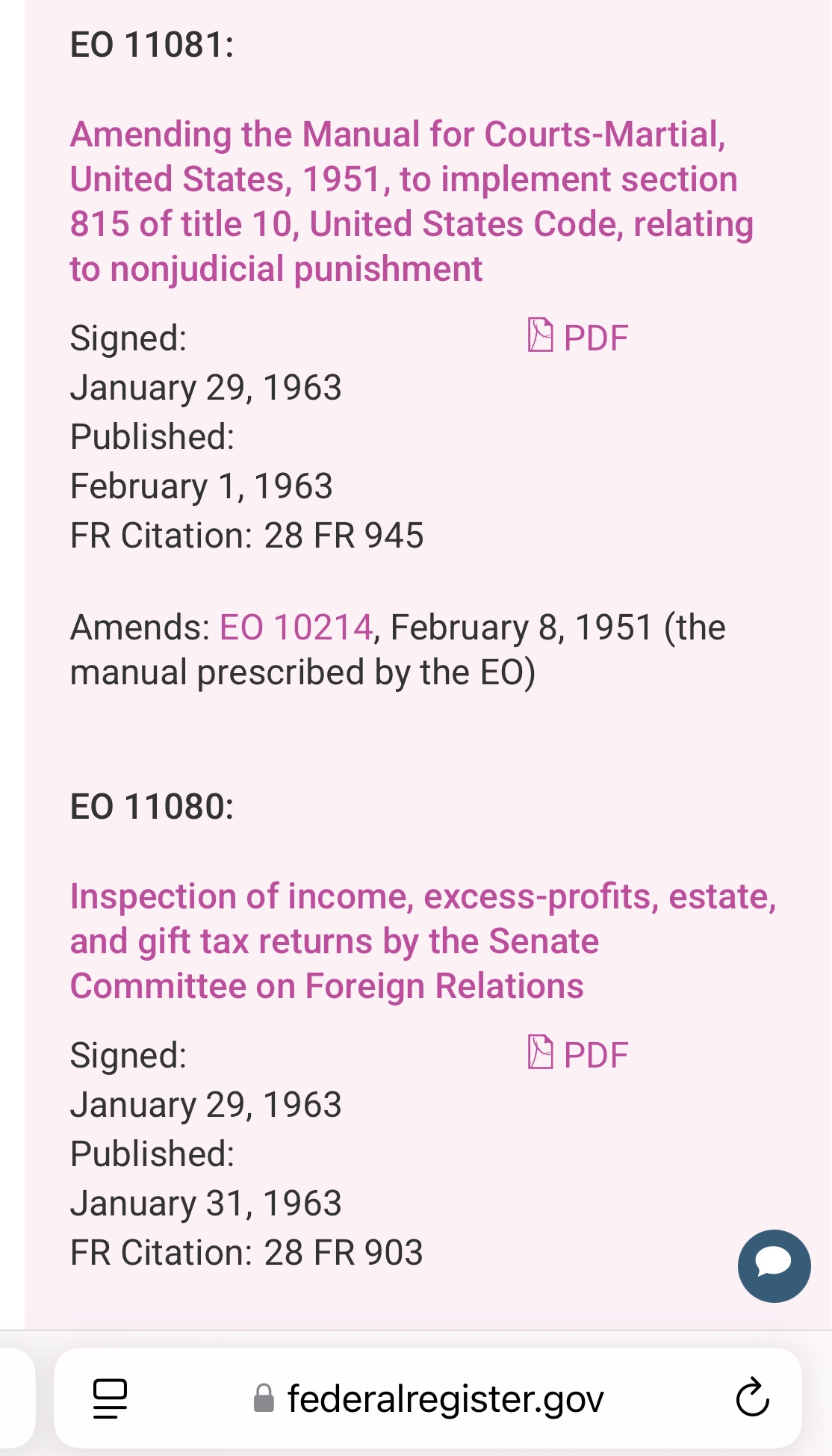
The line at the bottom of the diploma says: "This Doctorate is issued for the recipient's work on plasma and positrons pursuant to Executive Order APP#43 issued January 29, 1963 by John Fitzgerald Kennedy, President of the Unisted [sic] States of America." But the list of executive orders issued by JFK is public, and it doesn't include any order number "APP#43".
I tried to check if the diploma featured fonts which had not yet been published in 1974, but I believe the two fonts I was able to identify had both been published before 1974 (even though it's possible that Fleming would have used newer near-identical variants of the old fonts I identified, because there's newer copycat versions of both fonts, but I didn't bother doing a detailed investigation of the fonts).
The signatures of Francis F Chen and CD Anderson seem to have been written in some version of the Brush Script font, which was designed in 1942:
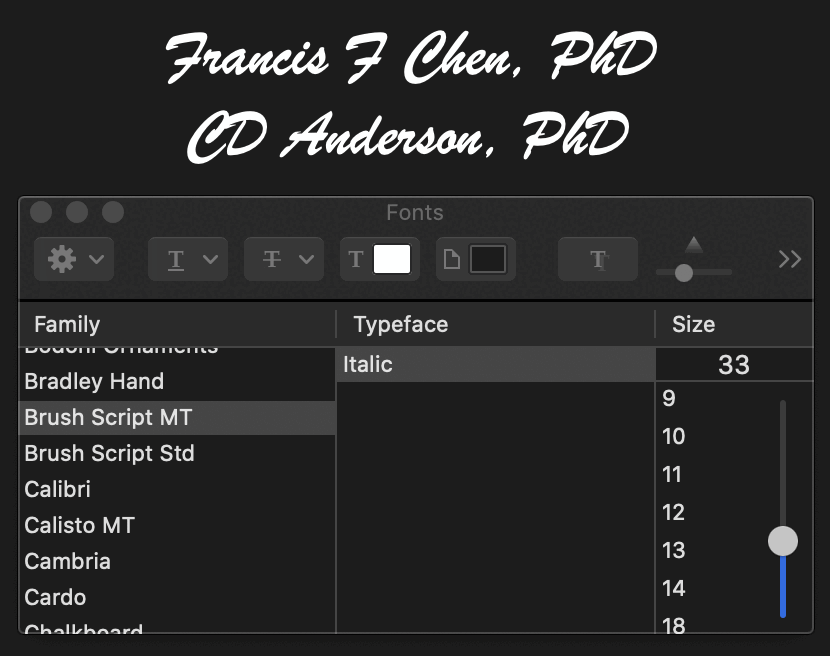
At first I thought the blackletter font in the diploma was Monotype Old English Text which was only published in 1990. [https://www.myfonts.com/collections/old-english-text-font-monotype-imaging] However Monotype Old English Text was a clone of an older font called Cloister Back that was designed around 1904, and based on the angle of the upper left arm in the capital letter U, Fleming's font actually seems closer to Cloister Black than to Monotype Old English: [http://identifont.com/show?249]

When I linked to the thread by LogarithmicDis, Fleming told me: "This image of my doctoral diploma is clearly altered and you are either guilty of altering it or guilty of presenting an altered diploma. It's a new country. :-)". [https://x.com/Doctor_I_am_The/status/1882159478986170631] Fleming also told me: "This doctorate was manipulated by people on line. People who spend a lot of time manipulating images including published research." [https://x.com/Doctor_I_am_The/status/1882153774636028052] However I didn't find any way the images in the thread by LogarithmicDis were manipulated apart from the adjustment of contrast which she mentioned in her thread.
But perhaps Fleming was projecting his own sins onto his adversaries, because at some point after LogarithmicDis posted her thread, Fleming altered the image of his diploma on his website so that the background of the signatures on the left and the two seals on the middle was modified to match the background of the rest of the paper: [https://www.flemingmethod.com/publications]

The old version of the image where the backgrounds didn't match is still available at the Wayback Machine: [http://web.archive.org/web/20230206233241/https://www.flemingmethod.com/publications, https://archive.is/Z1Ja4]

When I asked Fleming about his PhD thesis, he told me to read his book "Are We the Next Endangered Species". [https://x.com/Doctor_I_am_The/status/1878458620402151869] I believe he wanted me to read the appendix in his book called "Fleming Doctorate Research and Training". However I didn't find the book on Libgen or Anna's Archive, and it costs 27 USD on Amazon, but I don't want to pay that much for a single book chapter.
Fleming used to have a Twitter account called ExperiencedStud where he advertised his work as an actor and he tried to get casted in adult films. There's no snapshots of the account at the Wayback Machine, but Cheshire managed to take these screenshots of the account: [https://x.com/Thatsregrettab1/status/1559607005848883200/photo/1]
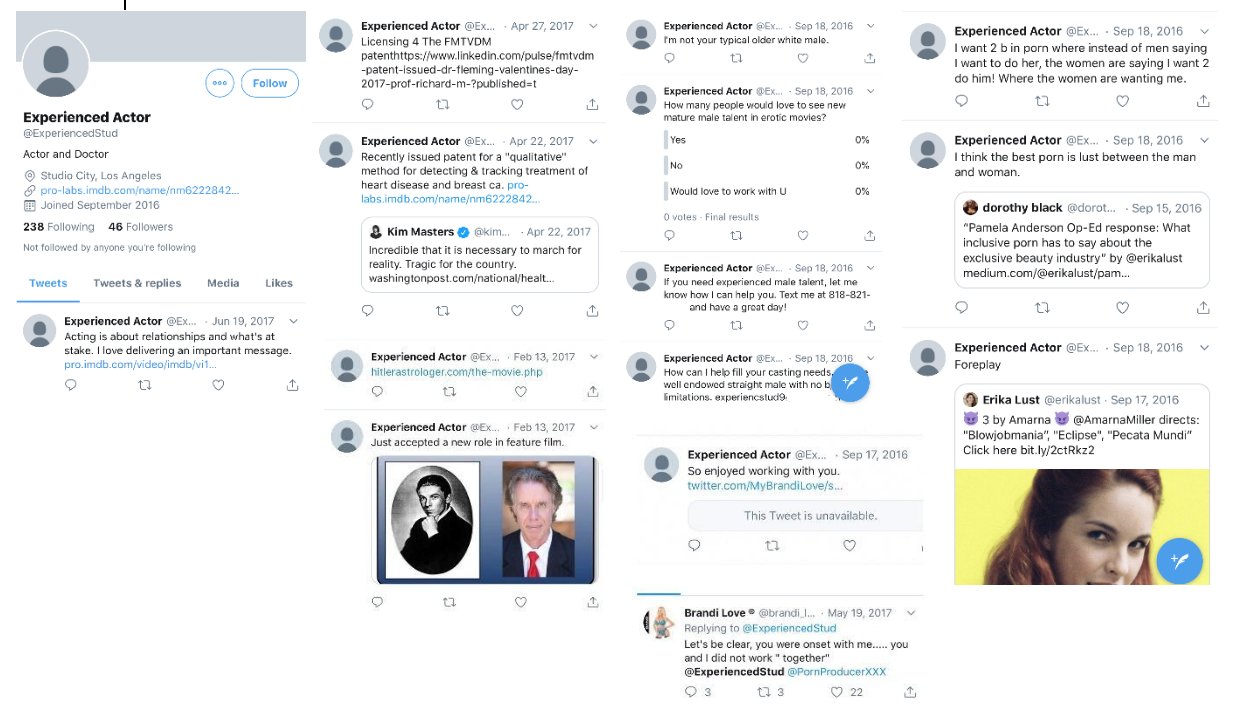
I don't know if Fleming actually managed to appear in any adult movie, because one of his tweets from 2016 said: "I've been acting a couple years. Just started in porn a few weeks ago after people told me for years I'm a natural. Looking to be your talent". [https://x.com/Thatsregrettab1/status/1386706893788258313]
Fleming said that the account was hacked, and that when people said he was an adult actor, it just meant that he was not a child actor: [https://x.com/Doctor_I_am_The/status/1386712519436349440, https://x.com/Doctor_I_am_The/status/1788273755619242277]
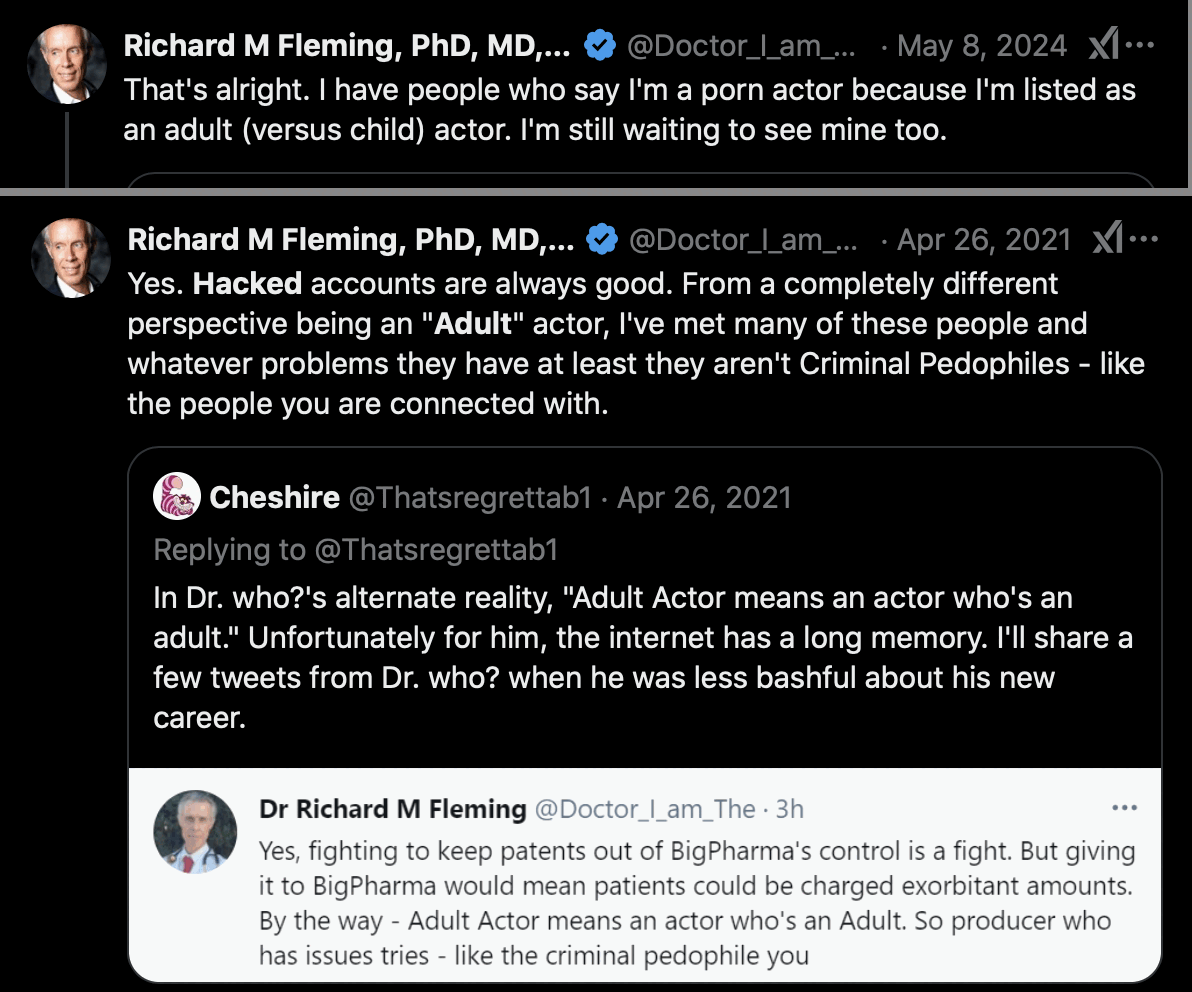
Fleming also claimed that the account was called "Experienced Student that got truncated and then hacked." [https://x.com/Doctor_I_am_The/status/1386723883647131658] (The truncation theory is actually somewhat plausible, because when you register a new account on Twitter, you are first asked to enter a display name, and then Twitter automatically suggests a username based on the display name which is truncated to at most 15 characters, and "ExperiencedStud" happens to be 15 characters long. But why would Fleming not have edited the username manually?)
However as evidence against Fleming's hacking theory, the court documents of the Fleming v. Sims case said: "According to Larry Sims, the parties first came into contact after Defendants placed a job posting on an adult entertainment job listing website. (See, e.g., ECF Nos. 90 at 1-2, 124 at 1, 188 at 1, 234 at 1, 244 at 1.) Plaintiff allegedly responded to the advertisement and represented himself as an adult film actor and model. (Id.)" [https://www.courtlistener.com/docket/6282193/247/fleming-v-sims/]
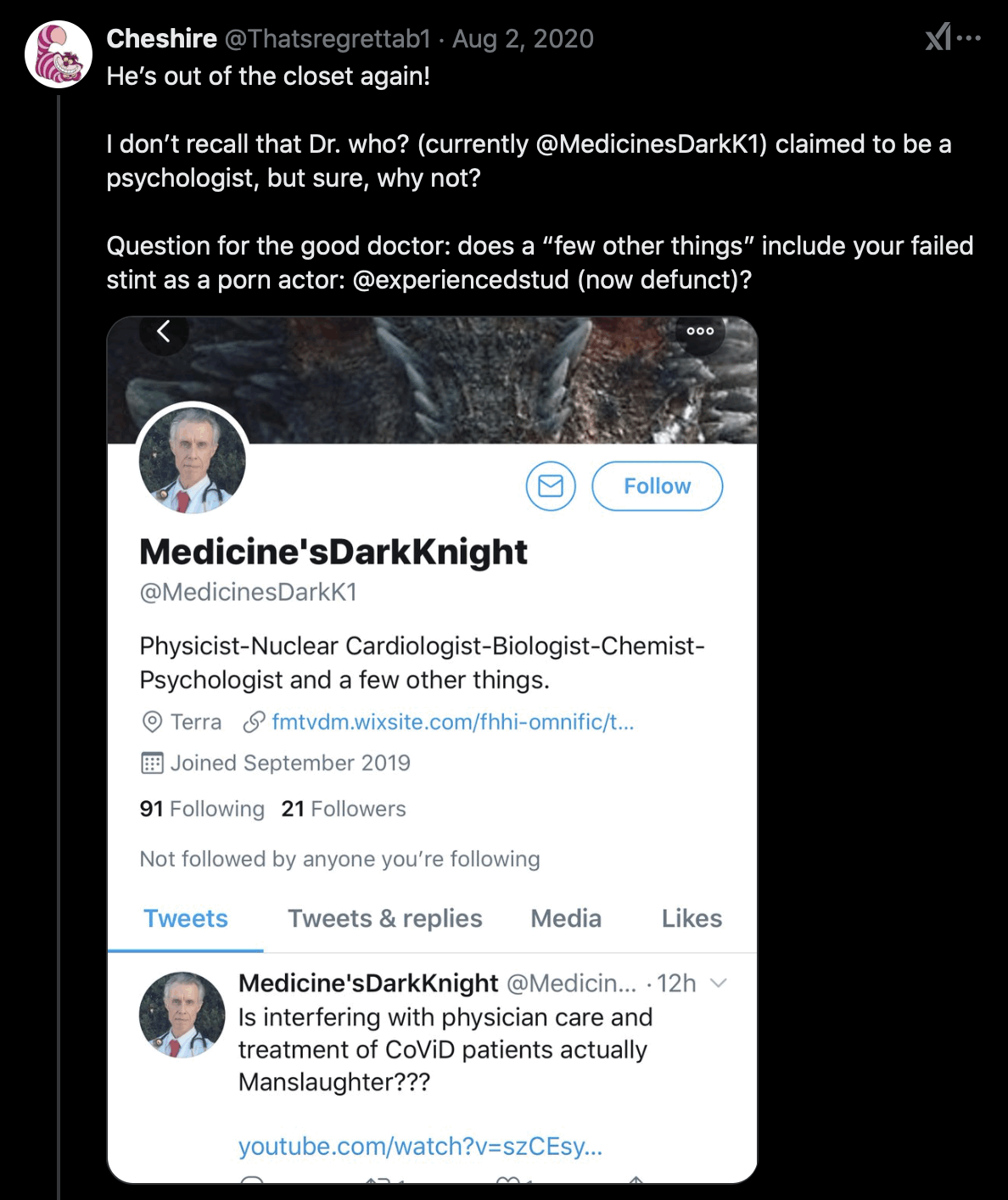
Fleming's current Twitter account used to be called MedicinesDarkK1 until October 2020. [https://x.com/search?q=medicinesdarkk1&f=live] After that he briefly changed his username to Lys_og_sannhet (which means "light and truth" in Norwegian) until he settled with his current username Doctor_I_am_The. [https://x.com/Thatsregrettab1/status/1331705114659229696] His old username was probably a reference to how Batman is called the Dark Knight: [https://x.com/Thatsregrettab1/status/1876688818805350811]
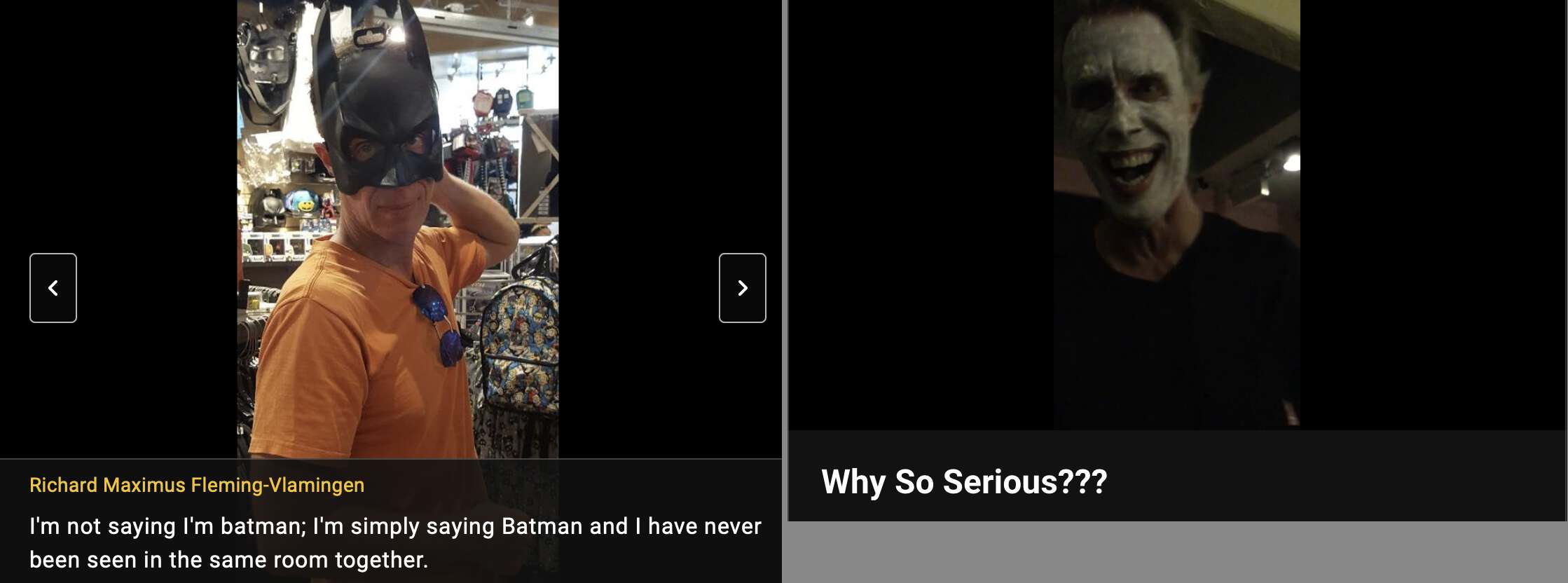
Fleming's IMDB profile has a photo where he poses as Batman and a video where he poses as Joker: [https://www.imdb.com/name/nm6222842/mediaviewer/rm57834240/, https://www.imdb.com/video/vi1525398297/]
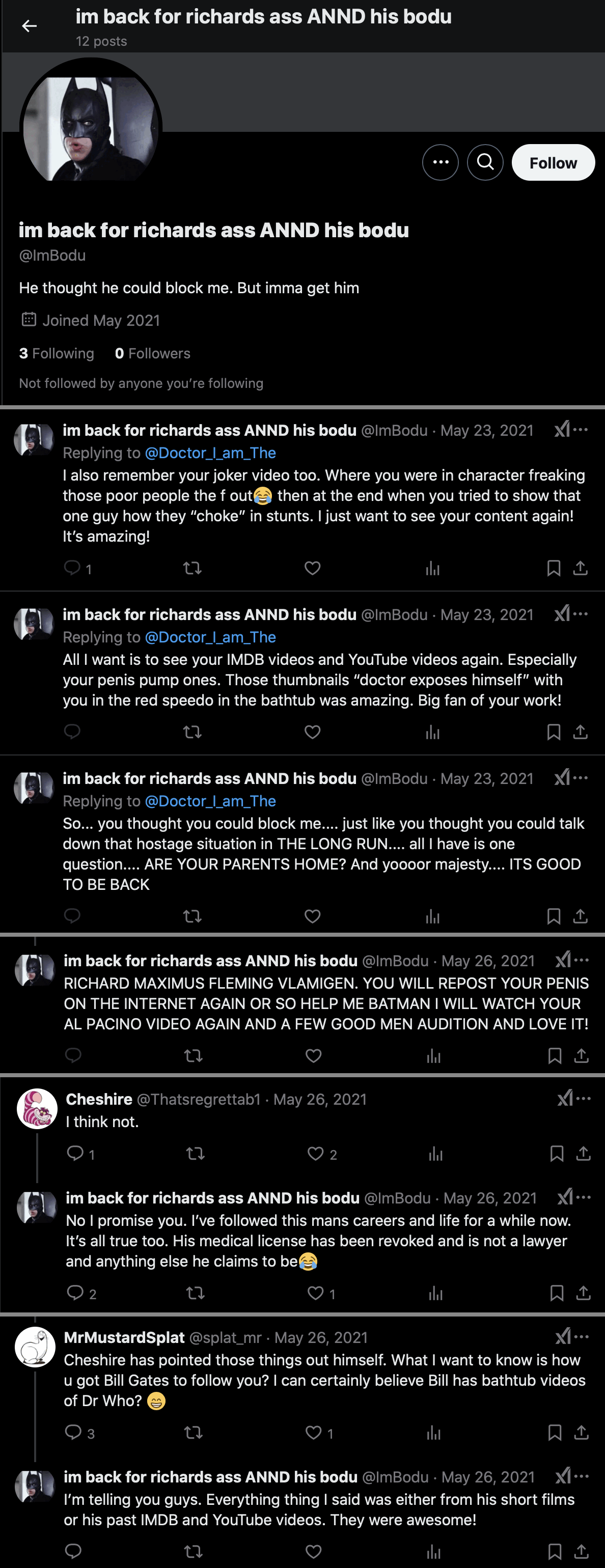
There's an account with a Batman avatar created in May 2021 called "ImBodu" (where the "Im" seems to match "I_Am" in the username of Fleming's main account). ImBodu currently only has 12 tweets. Its display name is "im back for richards ass ANND his bodu" and its bio says "He thought he could block me. But imma get him". Cheshire suspected the account might have been created by Fleming himself. [https://x.com/Thatsregrettab1/status/1397312313531650050] The account seemed familiar with several of Fleming's short films, his IMDB videos, and his YouTube videos, which likely few people besides Fleming himself would know about. They supposedly included a Joker video which would match the Batman theme, an audition video, a penis pump video, and a video where Fleming exposed himself in a speedo. The Batman account seems to have wanted Fleming to post his penis on the internet again (which likely few people besides Fleming himself would be interested in): [https://x.com/imbodu/with_replies, https://archive.is/molua]
In 2019 Fleming published a paper titled "Statistical Demonstration that FMTVDM is Superior to Mammography". [https://www.remedypublications.com/open-access/statistical-demonstration-that-fmtvdm-is-superior-to-mammography-5186.pdf] FMTVDM stands for the "Fleming Method for Tissue and Vascular Differentiation and Metabolism", which Fleming claims had a perfect accuracy of detecting breast cancer 1000 times out of 1000 when mammography detected breast cancer correctly only 828 times out of 1000:
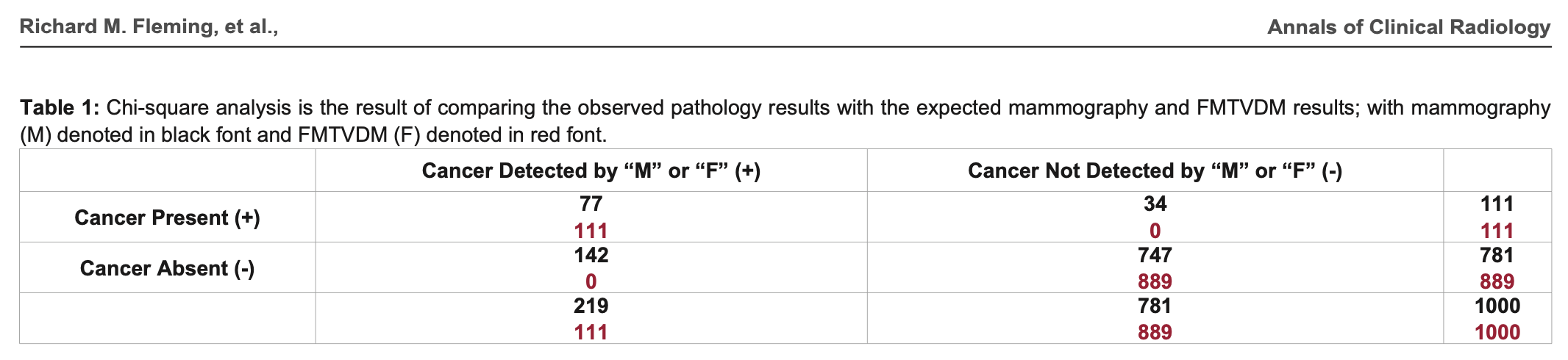
In the table above the sum on the second row is wrong, because
142+747 adds up to 889 and not 781. But that's the least of
Fleming's problems. The formula he used to calculate the chi-squared
test is also wrong, and it has two divisions by zero:

In his formula "χ2 = E (O-E)2/E", the first "E" should've been "∑" and the second number 2 should've been superscripted: "χ2 = ∑((O-E)2/E)".
Fleming also misunderstood the meaning of the expected counts E in the formula. He treated the results of FMTVDM as the expected counts and he compared it to the results of mammography which he treated as the observed counts. But his observed counts should've consisted of a contingency matrix with one row for mammography and another row for his Fleming method. And then he should've calculated the expected matrix based on the observed matrix by doing a matrix multiplication of the row sums and column sums and then dividing the result with the sum of all values in the contingency matrix:
> m=rbind(mammography=c(77,34,747,142),Fleming_FMTVDM=c(111,0,889,0))
> colnames(m)=c("present_correct","present_incorrect","absent_correct","absent_incorrect")
> m
present_correct present_incorrect absent_correct absent_incorrect
mammography 77 34 747 142
Fleming_FMTVDM 111 0 889 0
> expected=rowSums(m)%*%t(colSums(m))/sum(m)
> expected
present_correct present_incorrect absent_correct absent_incorrect
[1,] 94 17 818 71
[2,] 94 17 818 71
And only after that he should've calculated
sum((observed-expected)^2/expected):
> sum((m-expected)^2/expected) 194.4741 > chisq.test(m,correct=F) # same result Pearson's Chi-squared test data: m X-squared = 194.47, df = 3, p-value < 2.2e-16
Here the procedure of calculating the expected matrix is written out using basic arithmetic operators:
> rowsums=c(77+34+747+142,111+0+889+0)
> colsums=c(77+111,34+0,747+889,142+0)
> sum=c(77+111+34+0+747+889+142+0)
> rowsums
[1] 1000 1000
> colsums
[1] 188 34 1636 142
> sum
[1] 2000
> mult=matrix(c(1000*188,1000*34,1000*1636,1000*142,1000*188,1000*34,1000*1636,1000*142),byrow=T,ncol=4)
> mult
[,1] [,2] [,3] [,4]
[1,] 188000 34000 1636000 142000
[2,] 188000 34000 1636000 142000
> mult/sum # expected matrix
[,1] [,2] [,3] [,4]
[1,] 94 17 818 71
[2,] 94 17 818 71
I don't know if it would've been better to just use a simple 2 by 2 contingency matrix with one column for the number of correct detections and another column for the number of incorrect detections, but it gave me close to the same chi-squared value as the test with the 4 by 2 matrix:
> m=rbind(mammography=c(77+747,34+142),Fleming_FMTVDM=c(111+889,0+0))
> colnames(m)=c("correct","incorrect")
> m
correct incorrect
mammography 824 176
Fleming_FMTVDM 1000 0
> chisq.test(m,correct=F)
Pearson's Chi-squared test
data: m
X-squared = 192.98, df = 1, p-value < 2.2e-16
> expected=rowSums(m)%*%t(colSums(m))/sum(m)
> sum((m-expected)^2/expected)
[1] 192.9825
Fleming claimed that his Twitter account had been hacked so that someone had added a photo of David Icke to a poster for Fleming's Crimes Against Humanity Tour: [https://x.com/Doctor_I_am_The/status/1537897704830840836, https://archive.is/2BM0D]
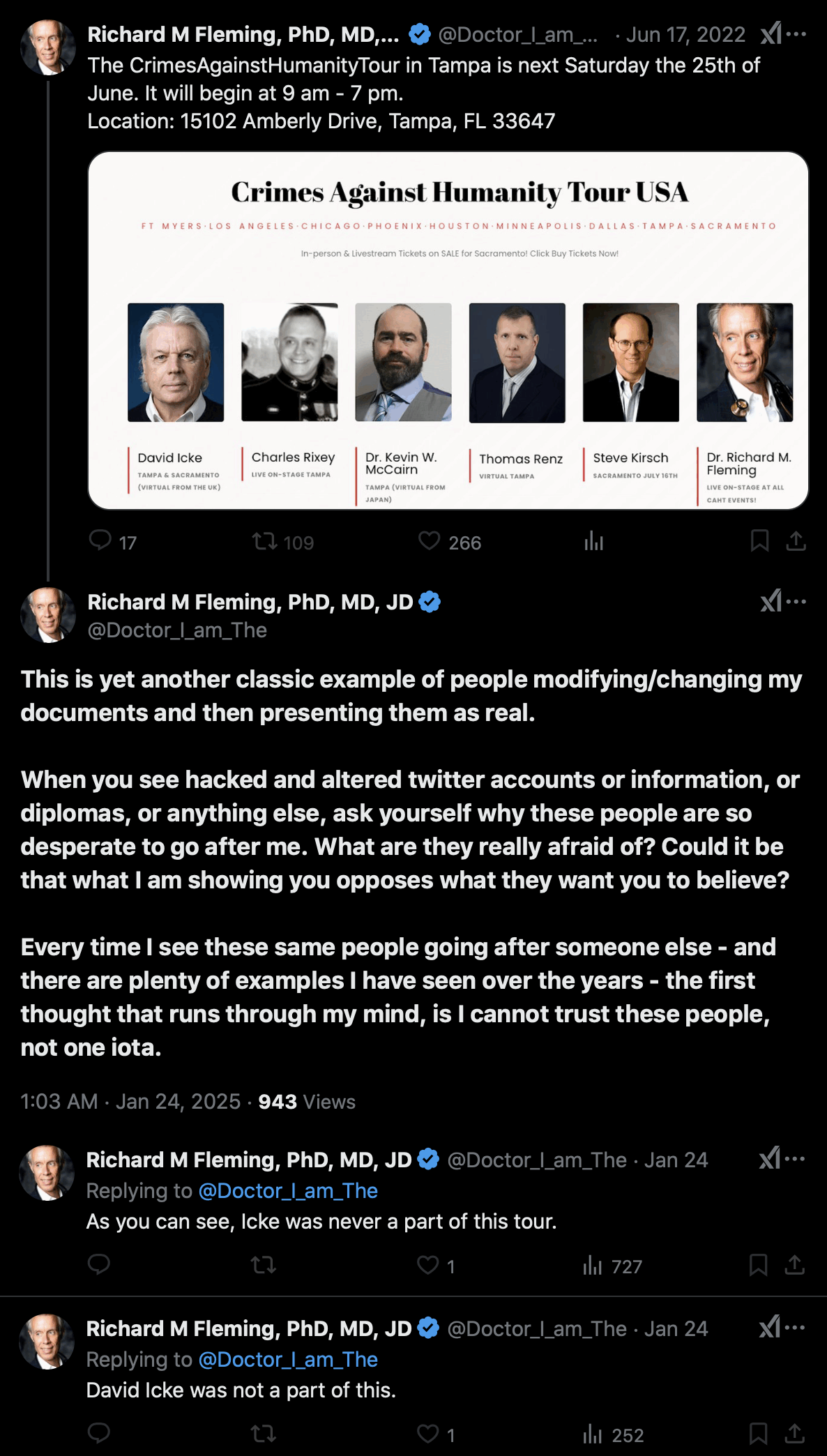
But then was Fleming's Facebook account also hacked? The Facebook account of the tour also has a poster that features David Icke: [https://www.facebook.com/103054418992983/photos/142901295008295/, https://archive.is/qGiFS]
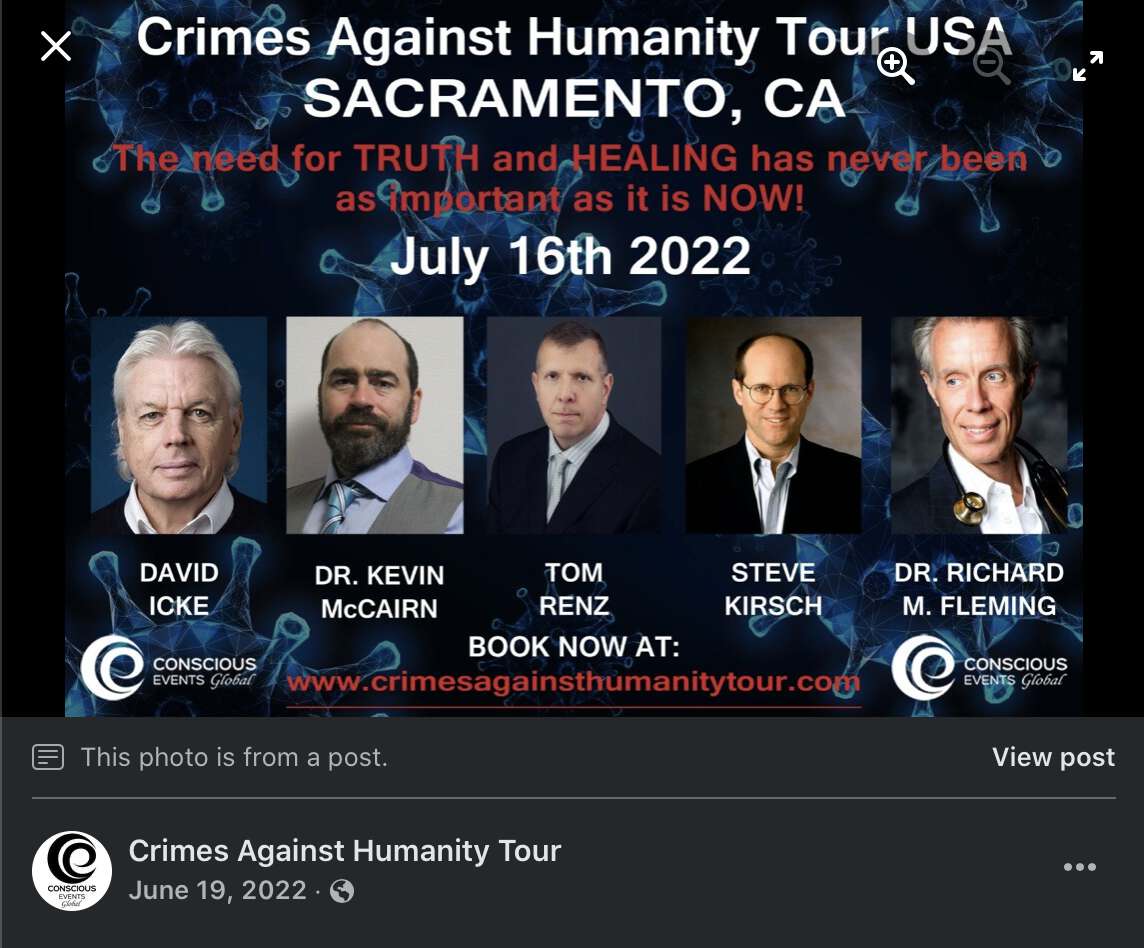
The tweet by Fleming that featured Icke was posted on June 17th 2022 UTC, and the Facebook post that featured Icke was posted on June 19th 2022 UTC.
David Icke's blog also has a post dated May 24th 2022 which says: "I am currently traveling the country with Drs. Judy Mikovits, Richard Fleming and Reiner Fuellmich. Our one-day conference topic in nine cities focuses on the case for crimes against humanity having been committed by leaders of Big Pharma and the biosecurity cartel." [https://davidicke.com/2022/05/24/monkeypox-technocracys-next-wave-of-crimes-against-humanity/]
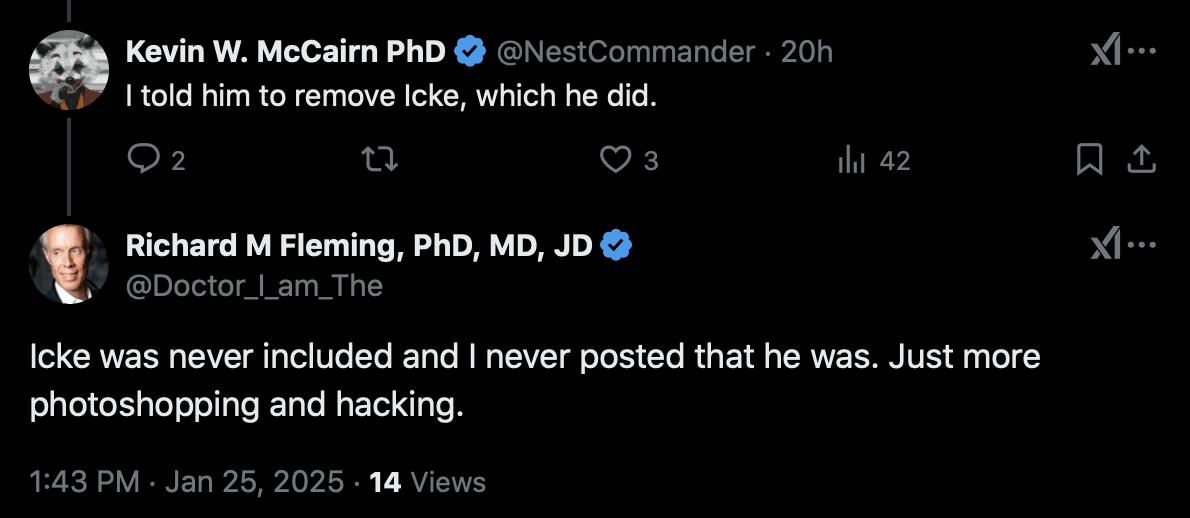
Kevin McCairn was also featured on the tour, and he told me that he asked Fleming to remove Icke. But even after that Fleming still denied that Icke was ever included on the tour and said it was just "more photoshopping and hacking": [https://x.com/NestCommander/status/1882872073883976179]
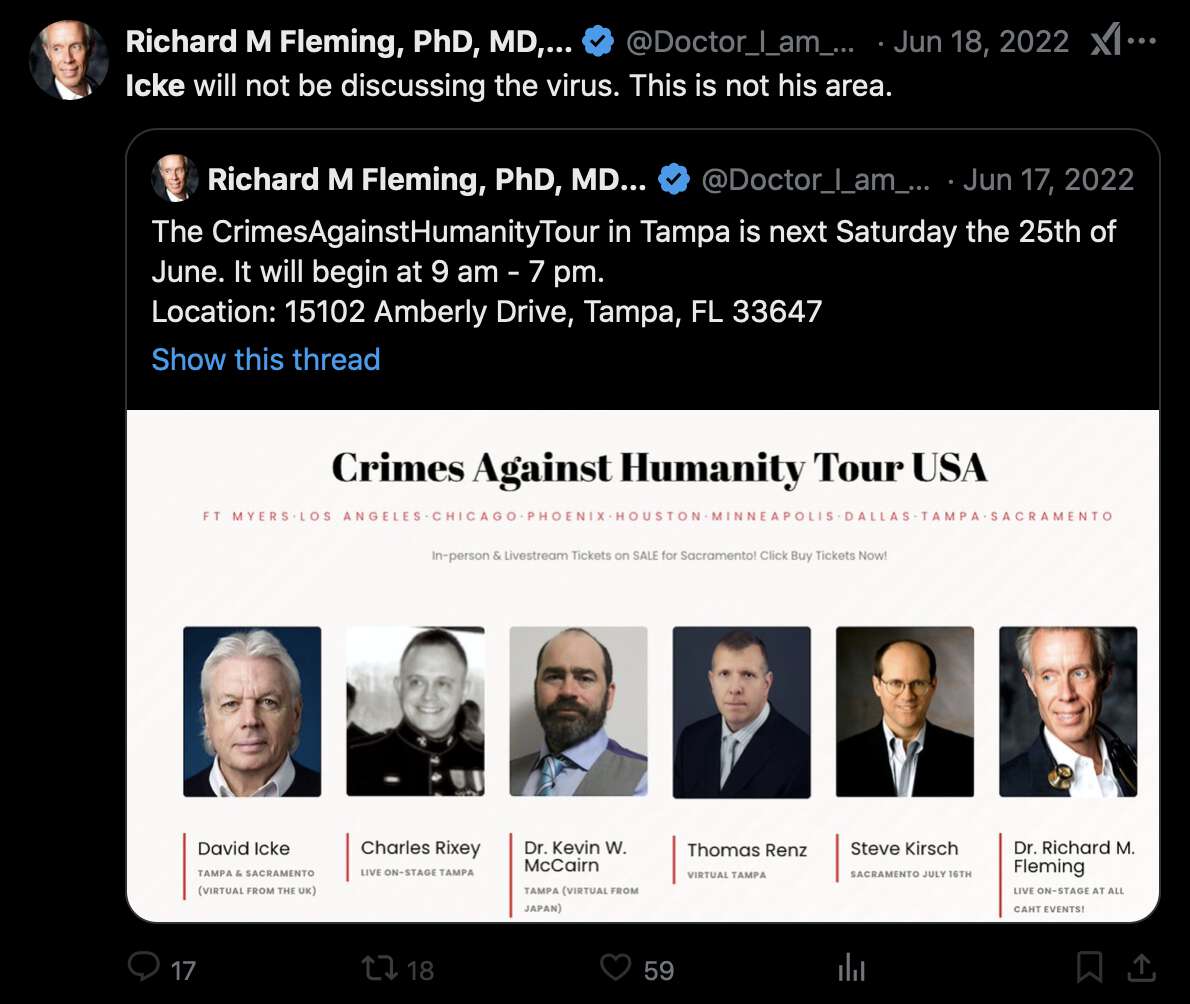
In 2022 Fleming even quoted the poster with Icke himself and referred to Icke in his tweet. So if the image file of the poster was photoshopped by the hackers, then did the hackers also edit Fleming's the text of Fleming's tweet so that it referred to the photoshopped poster? [https://x.com/Doctor_I_am_The/status/1537962322576125953]
In this section I'm mostly repeating the research of Cheshire. You can read his comments to this article first: https://retractionwatch.com/2020/05/29/a-convicted-felon-wants-people-to-enroll-in-a-covid-19-clinical-trial-what-could-go-wrong/.
Before the paper about Fleming's fake trial was published in a predatory journal, he published two preprint versions of the paper as well as two accompanying short reports. Here's links to all of them for the sake of convenience:
Cheshire posted this comment about the paper at PubPeer: [https://pubpeer.com/publications/618E865FABF6345892579A16F2AFEB]
Highlights: 23 sites, 7 countries, 1,800 patients, 10 treatment arms. No other authors other than Fleming and son. None of the sites were identified, although the ill-formatted tables (apparently due to unfamiliarity with Acrobat) include Table 3 showing the country of the sites. No funding was required for this study (?).
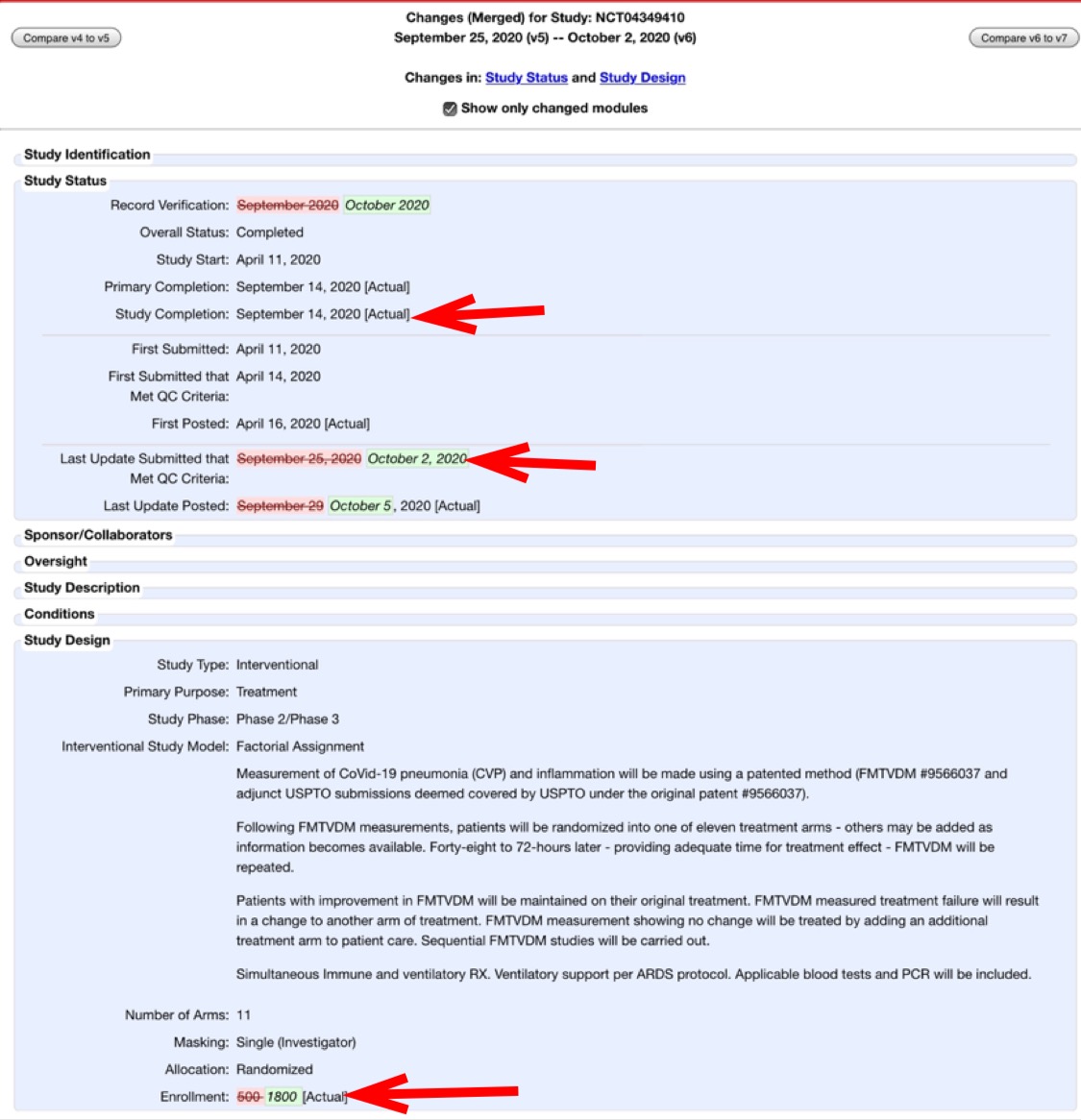
Interestingly, the Clinicaltrials.gov site shows that on October 8, the first author/sponsor changed the "treatment arms and interventions" details on all of the tested protocols. This is 3 weeks after the trial was marked concluded. An example of the changes:
Experimental: Treatment 1 ORIGINAL
Hydroxychloroquine 200 mg po q 8 hrs (600 mg qD) for a total of 10-days, and Azithromycin 500 mg IV on day 1, followed by 250 mg IV on days 2-5 (to prevent bacterial superinfection).
Experimental: Treatment 1 REVISED
Hydroxychloroquine 200 mg po q 8 hrs (600 mg qD) for a total of 10-days , OR Hydroxychloroquine 155 mg IV every 8-hours (600 mg qD) for 10-days if patient is intubated and Azithromycin 500 mg IV on day 1, followed by 250 mg IV on days 2-5 (to prevent bacterial superinfection). (emphasis added)
Source: https://clinicaltrials.gov/ct2/history/NCT04349410?A=6&B=7&C=merged#StudyPageTop
Other remarks:
- While there is mention of centralized institutional board review there is no mention of informed consent by patients or authorized representatives
- There does not seem to be any data provided as to the racial demographics of the patients
- Although 8 different Figures are referenced in the text, none appear in the preprint
- The author acknowledges 6 individuals (by initials only) for their assistance with the "individual patient centers." In comparison, a NEJM-published study on Remdesivir for COVID-19 with 35% fewer patients than this one, acknowledged over 200 physicians by name. https://www.nejm.org/doi/full/10.1056/NEJMoa2007764
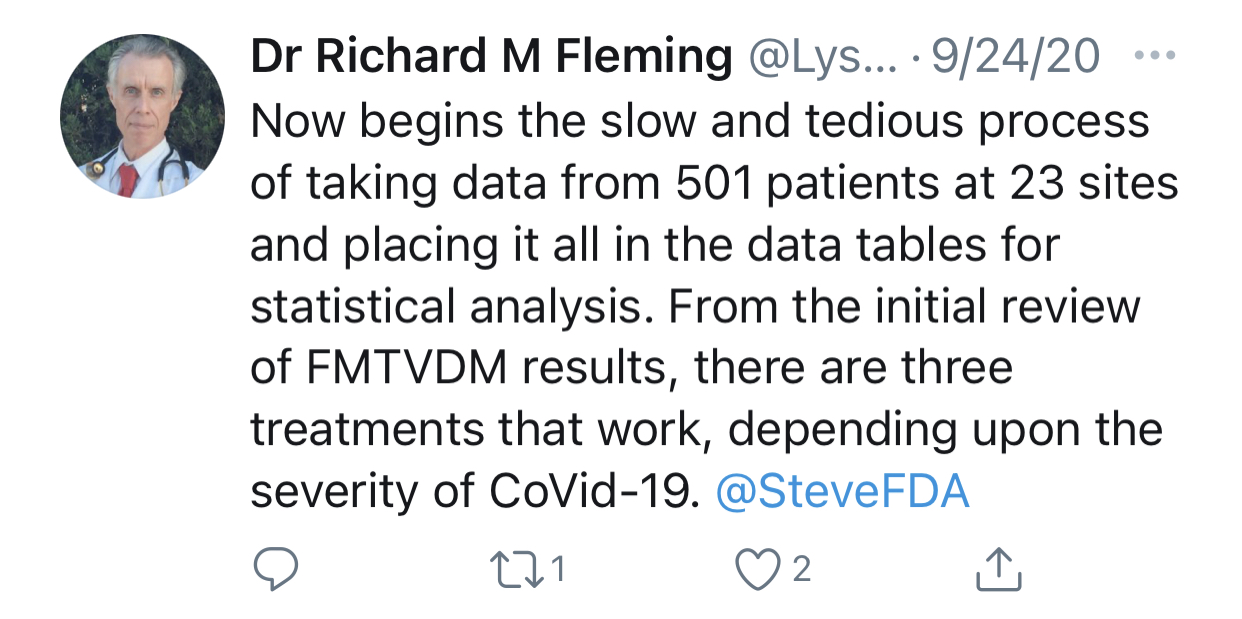
Cheshire also pointed out that after the trial is supposed to have already ended, Fleming changed the number of enrollees from 500 to 501 to 1,800:
Trial ended on September 14; at which time clinical trials.gov showed 500 enrollees.
Dr. Fleming posted this on Twitter on September 24, showing 501 enrollees:
On October 2, the sponsor (Dr. Fleming) changed the trial enrollment number to 1,800 at clinicaltrials.gov and 1,800 is the figure used in this preprint.
However the discrepancy seems to be because Table 3 shows that the total number of patients was 1,800, but 1,299 of them were outpatients and the number of inpatients enrolled to phases I and II was 501:
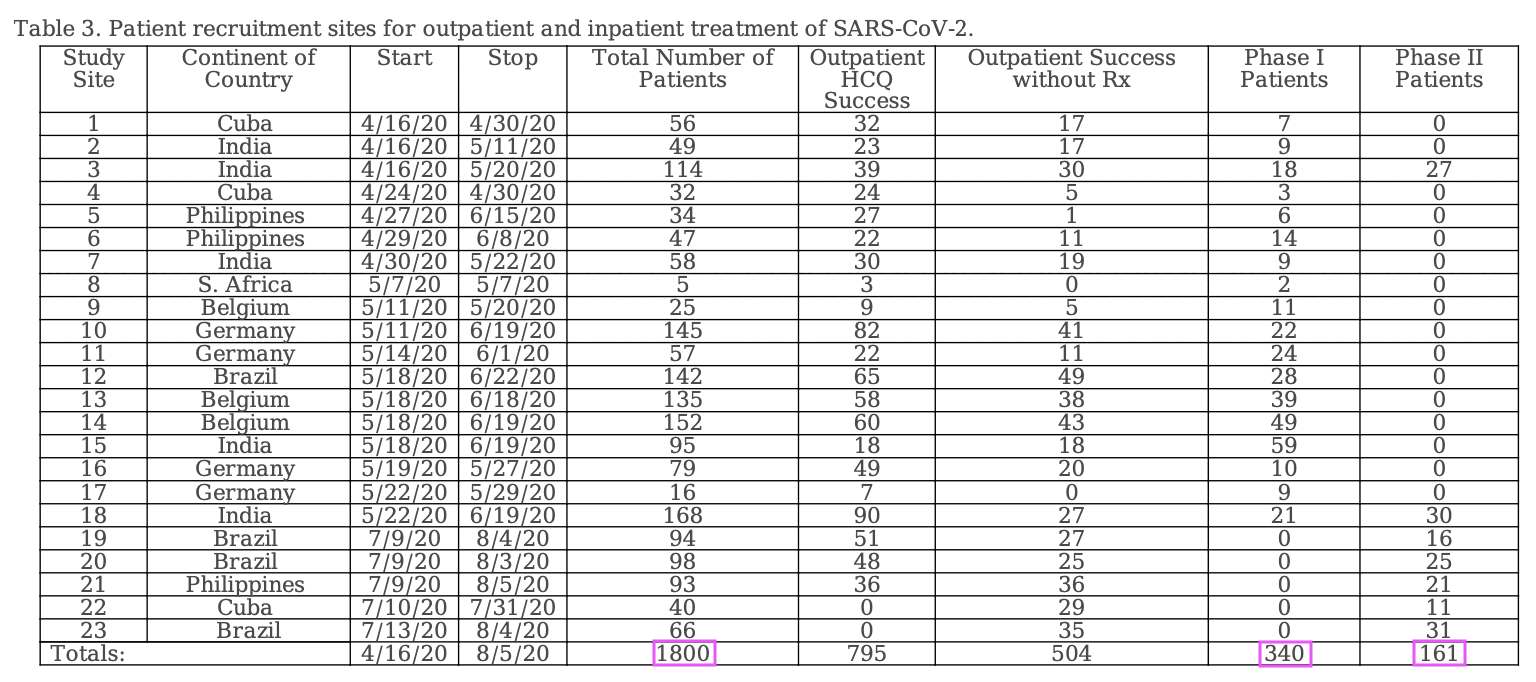
Fleming wrote that "NCT04349410 required identification of a seven-person SARS-CoV-2 treatment team" (where presumably each site would've had its own "treatment team" with at least seven persons, but the study is supposed to have included 23 sites so 23 times 7 would be 161). Cheshire wrote:
Given the extraordinary requirement that a 7-person treatment team (see below) be formed to treat the 501 hospitalized patients, how is it possible that all this additional care was provided with no funding from the sponsor? Why was this "required identification" not included as part of the pre-registered trial protocols? What evidence does the sponsor have that this "required identification" was followed in every case?

Someone also posted this comment at PubPeer:
It is noteworthy that these ambiguities about the study's feasibility and protocols are reminiscent of earlier skepticism, expressed 18 years ago, about another purported clinical trial from the same author:
...clinical trials are expensive, difficult and time-consuming. Even small dietary trials can easily cost several hundred thousand dollars and require entire research teams. The DPP estimated a cost of $1,075 just to recruit each participant. Fleming reports on a one-year trial of 100 participants and four diets with extensive follow-up. His paper, however, has no co-authors; it acknowledges no source of funding, nor any nurses, dietitians or technicians who might have helped. Fleming identifies himself as Medical Director of Preventive Cardiology, the Camelot Foundation at the Fleming Heart & Health Institute, but if his Web site or receptionist are any indication, he is the sole member of each of those.
As for the issue of peer-review, Fleming states that his patients "were randomly assigned to one of the four dietary regimens based upon dietary preferences." This protocol is pivotal to interpretation of the findings, yet oxymoronic: If patients were assigned to diets based on their dietary preferences, then they weren't randomly assigned. If they were randomly assigned, then their preferences must be irrelevant. The two methods are incompatible. If this paper was peer-reviewed, it was done poorly. If this constitutes high-quality research in this field, then I suggest even more skepticism is necessary.
source: https://www.washingtonpost.com/archive/lifestyle/wellness/2002/09/24/interactions/dfcd1470-eedb-474f-8ae8-714836e810d9/ [not behind paywall here: https://www.askbjoernhansen.com/2002/09/02/maybe_it_wasnt_a_big_fat_lie.html]
Cheshire also wrote:
This trial does not seem to have been registered in India as appears to be required:
My trial is already registered in another Primary Register, then why do I need to register again with the CTRI? A clinical trial being conducted in India, is also required to be registered in the CTRI as the CTRI captures data specific for the Indian arm of a trial, e.g. Site and PI details, Name of Ethics Committee and approval status, target sample size in India, start date in India etc.
Search here: http://ctri.nic.in/Clinicaltrials/advancesearchmain.php
Cheshire wrote: [https://pubpeer.com/publications/618E865FABF6345892579A16F2AFEB]
I'm confused again.
From the Methods section of this preprint (posted 10/27/20):
Outpatient Treatment: Patient recruitment for each outpatient treatment site is shown in Tables 3, 4 and Figure 2. Outpatient treatment was by definition provided by clinicians prior to hospital admission. Outpatient sites included private offices, physician and hospital clinics. Decision to treat (Treatments 1-4; Tables 2A and 2B) was made solely by the physician and patient. All outpatients received a minimum of 200 mg of elemental zinc daily while taking aminoquinolines. Following initial evaluation including PCR testing and initiation of treatment or the decision to provide no treatment, patients returned 3-5 days later for re-evaluation." (Emphasis added)
I'm puzzled how the treating physicians would have known to treat patients with 200 mg of zinc daily? In the protocol document dated April 2020 (still available at clinicaltrials.gov), there is no mention of pre-hospitalization or outpatient treatment. On July 4, 2020, an Appendix G was added that added a prehospitalization step which includes, "Begin immune supportive Rx, including Zn." No quantity of Zn is specified.
Further in a late September YouTube video by the first author he recommended 10 mg of daily zinc for prehospitalized COVID-19 patients.

If the protocol provided to physicians at the outset of the trial did not include prehospitalization instructions, but zinc (of unknown quantity) was included beginning in July, and in late September (after the conclusion of this study) the first author was recommending 10 mg daily Zn... how can the author make the statement that all outpatients received a minimum of 200 mg of elemental zinc daily?
Cheshire pointed out that a table in Fleming's preprint indicated that in all treatment groups patients received either 10 mg of zinc orally or 4 mg of zinc intravenously:
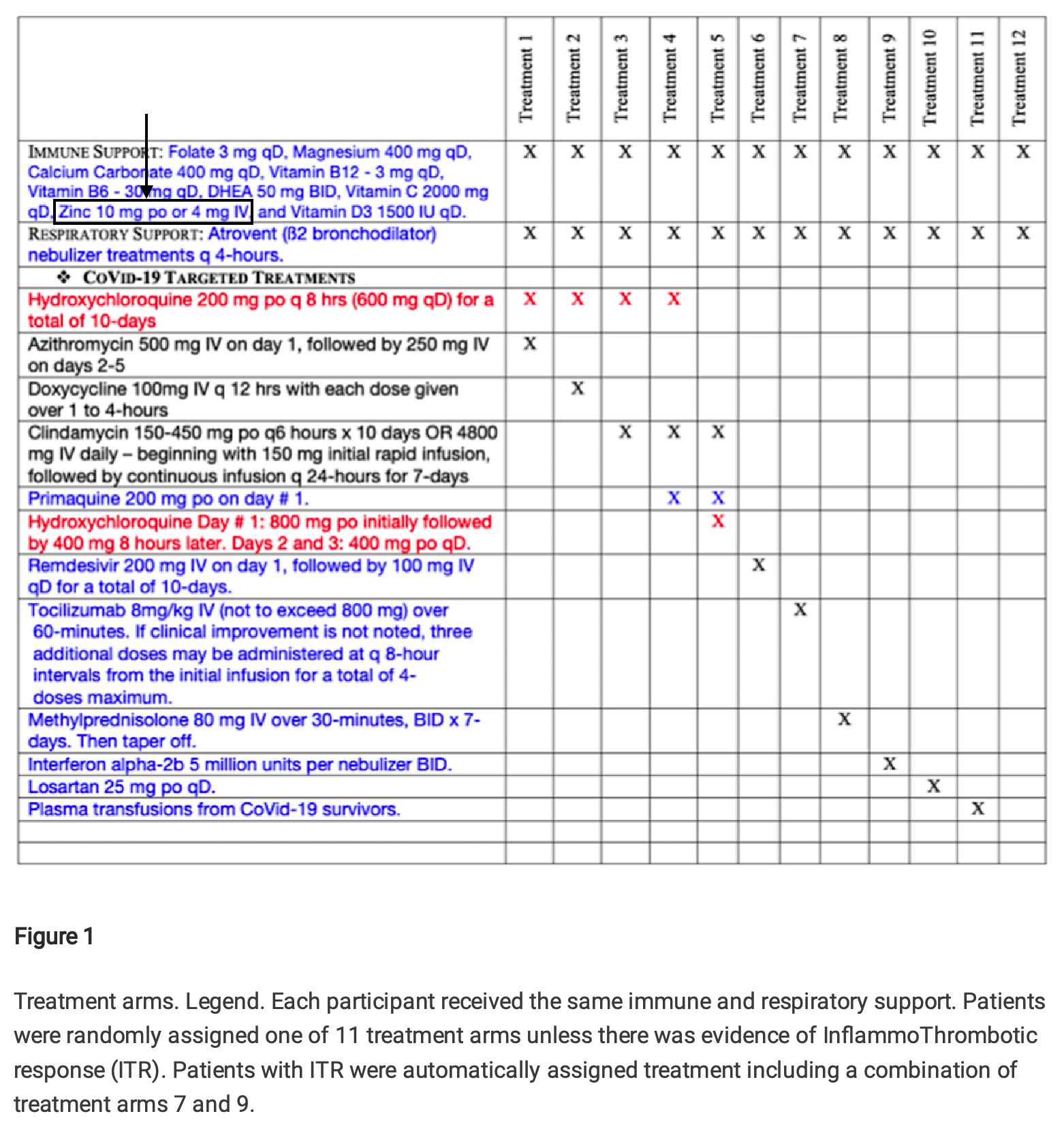
But Fleming responded by saying that his table showed the treatment regime in hospitalized patients and not outpatients:

However in case zinc was suspected to be helpful in treating COVID, why was the dosage of zinc reduced from 200 mg to 10 mg after a patient was hospitalized? In Fleming's YouTube video, he also recommended a dosage of only 10 mg of zinc to outpatients.
The abstract of Fleming's paper said: "The three successful treatment regimens include (1) Tocilizumab & Interferon a-2b, (2) Primaquine, Clindamycin, Tocilizumab & Interferon a-2b, and (3) Methylprednisolone. These three regimens were effective 99.83% of the time and shortened hospital stays from 40 ± 3 days to 1-2 weeks."
It's not clear how he determined the baseline hospital stay of 40 ± 3 days, because his study did not have any control group. I found no other reference to the figure of 40 ± 3 days apart from the abstract. The average duration of 40 days seems far too high, and the standard deviation of 3 days seems too narrow relative to the average duration.
A meta-analysis of COVID hospitalization studies from 2020 said: "We identified 52 studies, the majority from China (46/52). Median hospital LoS [length of stay] ranged from 4 to 53 days within China, and 4 to 21 days outside of China, across 45 studies." [https://link.springer.com/article/10.1186/s12916-020-01726-3] A meta-analysis from 2021 said: "The mean length of hospital stay due to SARS-CoV-2 infection was 12.5 days (SD 6.8)." [https://pmc.ncbi.nlm.nih.gov/articles/PMC8206636/] A study from a hospital in Brazil said that the mean duration of hospital stay was 10 ± 8 days. [https://www.sciencedirect.com/science/article/pii/S1876034122001563]
The conclusion of Fleming's paper said: "Successful treatment interventions focused on (1) avoiding intubation or extubating the patient within a matter of days - less than one week - to minimize the ARDS associated ventilator complications associated with the immunologic ITR to SARS-CoV-2, in addition to (2) using a combination of treatments within the first few days of admission including Interferon a-2b, Tocilizumab, and Methylprednisolone. These combinations were most effective if the patient had already received an aminoquinoline as an outpatient, or Primaquine as an inpatient. When provided the administration of convalescent plasma proved effective; however, given the limited supply of convalescent plasma, the potential consequences of a blood product transfusion including increased potential for thrombosis as a plasma product, and the availability of effective ITR treatments, convalescent plasma should be reserved for cases not responding to Interferon a-2b, Tocilizumab, Methylprednisolone, or the combination of Tocilizumab with Interferon a-2b. These ITR drugs proved most promising when initiated upon admission and when used in combination, reducing hospitalization time from 30-45 days to as little as 18-25 days with 0.17% mortality."
However 18 to 25 days is about 2.6 to 3.6 weeks, and not 1 to 2 weeks like Fleming wrote in the abstract.
The mortality rate of 0.17% seems to match the figure of 99.83% effectiveness Fleming mentioned in the abstract, so at first I thought that by effectiveness Fleming simply meant the percentage of subjects who did not die. Hovever then I thought I was wrong because I noticed that his paper said: "Successful treatment outcomes were defined using the quantitative measurements of FMTVDM with a reduction of ≥ 25, or a level of < 150, Ferritin levels < 270 ng/ml for men and < 160 ng/ml for women, and an IL-6 level of < 5 pg/ml." But next I noticed that Fleming also wrote: "Three hundred and forty patients entered Phase I and received sequentially added medical Treatment(s) until the patient demonstrated treatment success or expired." So apparently all of his imaginary subjects are supposed to have been treated until they either died or they recovered as measured by one of his three biomarkers, which explains why his efficacy against death is the same as his efficacy determined based on the biomarkers.
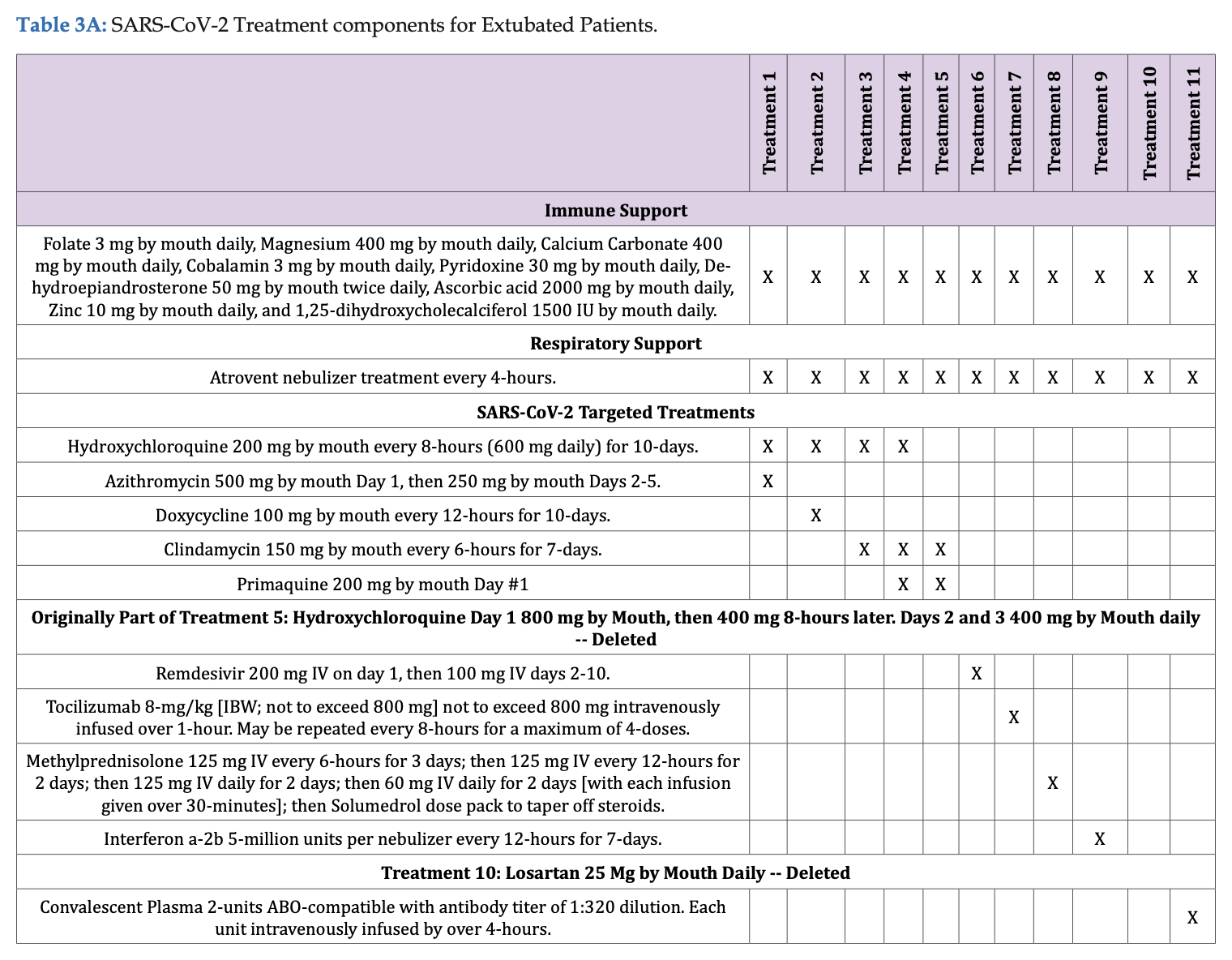
A mortality rate of 0.17% would correspond to 1 death per 570 to 606 people when rounded to the nearest digit. But I think the study didn't even have 570 subjects who received any of Fleming's so-called "three successful treatment regimes". There were only 501 inpatient subjects, and the section for outpatient treatment said: "Outpatient treatment was by definition provided by clinicians prior to hospital admission. Outpatient sites included private offices, physician and hospital clinics. Decision to treat (Treatments 1-4; Tables 3A & 3B) was made solely by the physician and patient." And Table 3A shows that none of Fleming's "three succesful treatment regimes" were given to outpatients, even though clindamycin alone was given to some outpatients (in the imaginary realm where Fleming conducted his trial):
Cheshire pointed out that people in all 11 treatment arms had the same baseline levels of ferritin and IL-6 at admission:
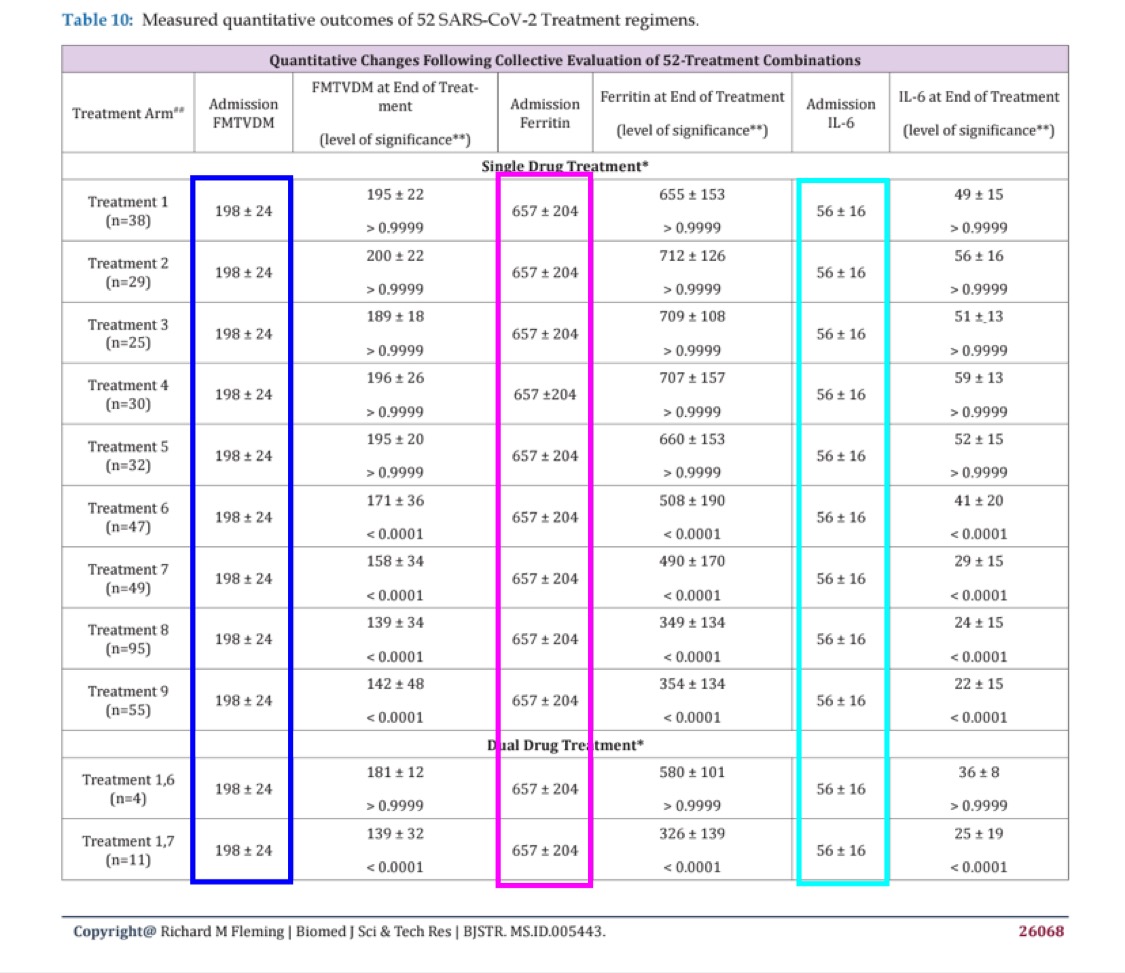
When I asked Fleming to explain it, he replied: "As explained in the paper, although clearly not read or comprehended by others, all participants began at baseline. This was baseline. They were then randomly assigned to treatments and then based upon measured FMTVDM outcomes, additional treatments were added or not. Statistical analysis was by multiple ANOVA." [https://x.com/Doctor_I_am_The/status/1878259706700984586] Then I asked him: "Ok, so did you first measure the ferritin level in each subject and then divided the subjects into treatment arms so that each arm happened to have the same average ferritin level with the same CI? It wouldn't be possible." (Where I should've said SD and not CI.) But Fleming replied: "You obviously are not very good at this and obviously have not read the paper. The sequence was laid out in the paper. You act like the typical student who has a reading assignment, shows up for class not having read the assignment and then try to blame the professor for your lack of understanding. Thus wasting your time, my time and everyone else's time."
But I still don't understand how it's possible for each arm to have the same levels of ferritin and IL-6 at admission.
Someone posted this comment: [https://retractionwatch.com/2020/05/29/a-convicted-felon-wants-people-to-enroll-in-a-covid-19-clinical-trial-what-could-go-wrong/]
According to the trial registration, "study chair" Fleming is conducting a clinical trial testing FDA regulated drugs for diagnosis and treatment in patients with Covid-19. Since there are no approved treatments for Covid-19, and IND application to FDA is required.
Per the FDA debarment notice, Fleming is prohibited "for 10 years from providing services in any capacity to a person that has an approved or pending drug product application."
https://www.govinfo.gov/app/details/FR-2018-09-28/2018-21210
Perhaps Retraction Watch could clarify with FDA that the debarment includes provision of services in FDA regulated research.
Cheshire posted this reply:
Source: https://www.healthgrades.com/media/english/pdf/sanctions/HGPYA59E0F8B7C744ECBB05212010.pdf
According to this State of Nebraska "Petition for Disciplinary Action," Fleming is "permanently excluded from Medicare, Medicaid, Tricare, and all other federal healthcare programs."
I verified that he is on the exclusion list accessible here: https://oig.hhs.gov/exclusions/index.asp and that site says:
OIG has the authority to exclude individuals and entities from Federally funded health care programs for a variety of reasons, including a conviction for Medicare or Medicaid fraud. Those that are excluded can receive no payment from Federal healthcare programs for any items or services they furnish, order, or prescribe. This includes those that provide health benefits funded directly or indirectly by the United States (other than the Federal Employees Health Benefits Plan).
OIG maintains a list of all currently excluded individuals and entities called the List of Excluded Individuals/Entities (LEIE). Anyone who hires an individual or entity on the LEIE may be subject to civil monetary penalties (CMP). To avoid CMP liability, health care entities should routinely check the list to ensure that new hires and current employees are not on it.
Based on this, it sounds unlikely that Fleming/Dr. who? would be in a position to legally run a clinical trial.
However I don't know to what extent US regulations would've applied to Fleming's trial since none of his imaginary trial sites were located in the United States. There were 5 sites in India, 4 in Germany, 4 in Brazil, 3 in Philippines, 3 in Cuba, 3 in Belgium, and 1 in South Africa.
A blog post about the journal where Fleming's paper was published said the following: [http://flakyj.blogspot.com/2019/11/biomedical-journal-of-scientific.html]
Although the ISSN entry for the Biomedical Journal of Scientific & Technical Research identifies it as a US publication, its mangled management of the English language suggests otherwise. The website describes the goal of this journal's "publishers" as follows:
The only motto of Biomedical Journal of Scientific & Technical Research (BJSTR) Publishers is accelerating the scientific and technical research papers, considering the importance of technology and the human health in the advanced levels and several emergency medical and clinical issues associated with it, the key attention is given towards biomedical research. Thus, asserting the requirement of a common evoked and enriched information sharing platform for the craving readers.
BJSTR is such a unique platform to accumulate and publicize scientific knowledge on science and related discipline. This multidisciplinary open access publisher is rendering a global podium for the professors, academicians, researchers and students of the relevant disciplines to share their scientific excellence in the form of an original research article, review article, case reports, short communication, e-books, video articles, etc.
BJSTR Publishers are self supporting, with no dependency on any other external sources (like universities, centers) for funds and strives for the best and enhanced quality publications competes the world wide open access publishing market.
We always rely on the support from the members of our BJSTR family that is relevantly our Authors, Editorial Committee members, advisory board, Reviewers Board and all the technical support teams all over the globe. We trust in the reciprocated coordination and cooperation in terms of sharing the scientific knowledge of individuals and Groups of Research centers/areas will in turn educates and provokes in advanced researches. In this case we would like to act as a media that anchors in the transformation of information in the form of global online publication.
With writing like this, the absence of "dependency on any ... external sources (like universities, centers) for fund" is understandable. So is the fact that the journal is not in the National Library of Medicine collection and not indexed for Medline.
When one researcher "submitted a string of machine-generated nonsense" entitled "The Expression of the Proper-Name Effect Reinforces the Disarticulation ofCommunicative [sic] Rationality," the article was "quickly accepted after scrupulous peer review." Evidently, the editors did not notice that the author was Harold E. Potter of the Institute of Improbabilistics, University of Bogus, UK. An author posting on Researchgate reports that when he declined to pay the $600 fee, the journal dropped the price to $99.
Wisely, the publishers do not reveal their identities. The domain name biomed-res.org in the email is registered to a privacy service. The contact information on the website is Biomedical Research Network+, LLC, 1 Westbrook Corporate Center, Suite 300, one Westchester, IL 60154, USA +1 (720) 414-3554 Fax - (720) 367-5187 info@biomedres.us angelaroy@biomedres.us. The registrant of biomedres.us provides the same physical address along with the name Biomedical Research Network + LLC and email URL of openaccessnetworks@gmail.com. The Westbrook Corporate Center is a property with "virtual office" space on sale for as little as $2 a day.
Someone posted this comment to the blog post: "ALL these BJSTR, IRIS, CRIMSON, LUPINE are interconnected
to SRAWIK GROUP a proprietor company in Hyderabad India". However
I'm not sure if it's correct or not because there were no other hits on
Google when I searched for
srawik "biomedical journal of scientific".
The associate editor of the BJSTR journal is suposed to be someone called "Angela Roy". Her location at Twitter is listed as "600 Third Avenue, 2nd floor, New York - 10016, USA". [https://x.com/Biomedres01] Her bio at Orcid doesn't say anything about herself or list any of her publications, but it only describes BJSTR in broken English: "Biomedical Journal of Scientific & Technical Research (BJSTR) is a multidisciplinary, scholarly Open Access publisher focused on Genetic, Biomedical and Remedial missions in relation with Technical Knowledge as well. We crave to select ground-breaking research based on modernism, aptness, scientific connotation, prospective spectator's interests, etc. We endeavor to provide by far and liberally accessible belvedere to researchers and practitioners in support of their novel and valuable ideas. We already have 2000+ Editorial Board members along with 5000+ Published articles in our box. Our BJSTR maintains a scrupulous, methodical, fair peer review System. Besides, quality control is riveted in each step of the publication process." [https://orcid.org/0000-0002-4278-5729]
This section addresses the PDF linked at the top of this page: https://www.flemingmethod.com/crimesagainsthumaniy. (Archive link: http://web.archive.org/web/20240217163948/https://www.flemingmethod.com/_files/ugd/659775_6532f42cb0b24f92bb36e224b7abac81.pdf.)
Fleming wrote: "When the EUA documents were used for the statistical analysis of the Pfizer, Moderna, and Janssen Drug Vaccine Biologics, and the Chi-Square analysis of the results published in those EUA documents was analyzed for the Pfizer vaccine as shown in the following graphic, there was no statistical difference between vaccinated and un-vaccinated people diagnosed with having COVID-19. To be statistically different (a benefit for people being vaccinated) the 'p (probability)- value' must be less than or equal to less than 5 times per hundred people. This is the scientific definition of statistical benefit and is written as 'p<0.05'. In the graphic the p-value was 0.224418 [note: actually 0.244218] and is NOT statistically significant; i.e. there is no statistical difference in the number of people diagnosed with COVID who were vaccinated when compared with the non-vaccinated group of people." And next he showed this image, where he made the images on the right himself but he took the table on the left from a review memorandum for the emergency use authorization of the Pfizer vaccine: [https://www.fda.gov/media/144416/download]

There were 8 cases out of 17,411 people in the vaccine group and 162 cases out of 17,511 people in the placebo group, so common sense should've dictated that the difference between the groups was statistically significant, and Fleming should've noticed that there was something wrong with his calculation because he only got a p-value of about 0.24.
In his contingency matrix the "observed"
column shows the number of people with no case, or for example
17411-8 = 17403 for Pfizer. However I don't understand how
he calculated the "expected" column. The
numbers in parentheses show the result of
rowSums(m)%*%t(colSums(m))/sum(m) (for example
34652*34752/69503 = 17326.25 for the top left square).
However the proper way to make the contingency matrix would've been to have one column for the number of people with a COVID case and another column for the number of people with no COVID case. It gave me a p-value of about 9e-32 here:
> cases=c(8,162);people=c(17411,17511)
> m=cbind(case=cases,no_case=people-cases)
> rownames(m)=c("Pfizer","placebo")
> m
case no_case
Pfizer 8 17403
placebo 162 17349
> chisq.test(m) # p-value shown as `<2.2e-16` due to limit of floating point precision
Pearson's Chi-squared test with Yates' continuity correction
data: m
X-squared = 137.5, df = 1, p-value < 2.2e-16
> .Machine$double.eps # smallest positive float `x` where `1+x!=1`
# [1] 2.220446e-16
> chisq.test(m)$p.value # p-value is much lower than 2.2e-16
[1] 9.395097e-32
> expected=rowSums(m)%*%t(colSums(m))/sum(m)
> expected
case no_case
[1,] 84.7566 17326.24
[2,] 85.2434 17425.76
> sum((m-expected)^2/expected) # manual calculation without correction
[1] 139.3046
> chisq.test(m,correct=F)$stat # matches manual calculation
X-squared
139.3046
Kevin McCairn also got a similar p-value when he used MATLAB to do a
chi-squared test (but the reason why McCairn said he got a lower p-value
than me was because my p-value was shown as < 2.2e-16 by
R even though it was actually about 9e-32 which was similar to McCairn's
result). McCairn wrote:
[https://x.com/NestCommander/status/1882185862257242263]
Richard I'm not following the logic, not that I trust Pfizer data, but the appropriate use of the Chi-squared in the tables above would be calculated as follows (MATLAB code below), I get a smaller P Value than Henjin, but I did no post-hoc corrections as it's only a 2x2 contingency table, and the result is highly significant.
% Data observed = [8, 162; 17403, 17349]; % Observed frequencies total = sum(observed, 'all'); row_totals = sum(observed, 2); col_totals = sum(observed, 1); % Expected frequencies expected = (row_totals * col_totals) / total; % Chi-squared calculation chi_squared = sum((observed - expected).^2 ./ expected, 'all'); % Degrees of freedom df = (size(observed, 1) - 1) * (size(observed, 2) - 1); % P-value calculation p_value = 1 - chi2cdf(chi_squared, df); % Critical value for p = 0.05 critical_value = chi2inv(0.95, df); % Display results fprintf('Chi-squared statistic: %.2f\n', chi_squared); fprintf('Degrees of freedom: %d\n', df); fprintf('Critical value (p=0.05): %.2f\n', critical_value); fprintf('P-value: %g\n', p_value); % Use %g for full precision scientific notation % Conclusion if chi_squared > critical_value disp('The result is significant at p = 0.05.'); else disp('The result is not significant at p = 0.05.'); end The result is significant at p = 0.05 χ2=139.3046 P=7.89×10−32
Fleming wrote "No other Coronavirus contains a Furin Cleavage site as shown in the following graphic":

But his graphic only contained sarbecoviruses. And there's sequences
of feline infectious peritonitis virus which contain an FCS at the S1/S2
junction with PRRAR:

Many merbecoviruses like MERS and HKU5 also have a furin cleavage site at the S1/S2 junction. The closest relative of sarbecoviruses are hibecoviruses, which are a small subgenus of betacoronaviruses that I believe currently includes only 4 published virus sequences (which are Hp-betacoronavirus/Zhejiang2013, Zaria bat coronavirus ZBCoV, CD35, and CD36). But all of them except ZBCoV have an FCS at the S1/S2 junction:

Fleming wrote: "The odds of such a cleavage site occurring spontaneously (naturally) is 3.21 x 10-11. [...] Simply put, the Furin cleavage site 'critical' for the SARS-CoV-2 viruses' entry into human cells resulting in disease and death, is astronomical. It has it not found in any other coronavirus at the critical S1/S2 location, and the US Government which has funded the Gain-of-Function research, owns the patent for this Furin Protease Cleavage Site, also associated with the HIV glycoprotein (HIV gp120) 120 (sialic acid raft receptor) and cancer progression."
The figure of 3.21e-11 comes from a paper by Ambati et al. titled "MSH3 Homology and Potential Recombination Link to SARS-CoV-2 Furin Cleavage Site". [https://frontiersin.org/journals/virology/articles/10.3389/fviro.2022.834808/full] But in the paper 3.21e-11 was not calculated to be the odds of an FCS occurring naturally but the odds that a given 19-base sequence would be contained both within a random 30,000-base sequence and within a set of 24,712 random 3,300-base sequences.
The paper by Ambati et al. included the image below with this caption: "Calculations of the probability of natural occurrence of the 19nt sequence under study. The SARS-CoV-2 genome is ~30,000 nucleotides long (P1). The patented sequence is ~3,300 nucleotides long (P2). The patented library encompasses 24'712 sequences of varying lengths with median length being in the range of 3,300 nucleotides. Conventional probability calculations are given of the probability of the presence of a 19-nucleotide sequence in the human genome and in one of the patented library sequences."

Ambati calculated the likelihood that a given 19-base segment would
be included in SARS-CoV-2, and he multiplied it with the likelihood that
the same segment would be included in the single Moderna patent happened
to match the 19-base segment. But he should've instead just calculated
the likelihood that a given 19-base segment would match the Moderna
patent by chance, like what he did in his P2c
probability.
In order to search the patent BLAST database for sequences that were
published before 2020 and that matched the keyword "Moderna", I used the Entrez query
0[0:2019] Moderna. It returned a total of 669,810 sequences
with a total length of about 1,011,731,339 bases. So if all sequences
are assumed to be random, then the likelihood that any of them would
match a given 19-base segment would be about 0.007:
1-((1-.25^19)^(1e9*2)) (where the likelihood of one or more
matches is calculated by subtracting the likelihood of zero matches from
1, and 1-.25^19 is the likelihood that a single 19-base
segment segment doesn't match the target sequence, and
1e9*2 is the approximate number of 19-base segments on both
strands in all Moderna patent sequences). But since there's 8 different
ways to choose a 19-base segment of SARS-CoV-2 that contains the FCS
insert and a total of 7 surrounding bases from either side combined, the
likelihood further increases to 1-((1-.25^19)^(1e9*2*8))
which is about 0.06.
For example if you pick two sets of 10 unique random integers between
1 and 1000, then what is the likelihood that both sets include one or
more shared integer? Ambati's logic for calculating the probability of
3.21e-11 was equivalent to calculating the likelihood that a given
integer like 123 would be incluced in both sets, which is
(10/1000)*(10/1000). But the real likelihood that the two
sets contain one or more shared integers is
1-kcombinations(990,10)/kcombinations(1000,10), which is
about 0.10 (where kcombinations(990,10) is the number of
ways to choose 10 unique integers which don't match any of the 10 unique
integers in the other set).
Ambati's probability calculation also had other more minor issues which were mentioned in a response to his paper that was later published in the same journal: [https://www.frontiersin.org/journals/virology/articles/10.3389/fviro.2022.914888/full]
P2c probability indicated the likelihood that
a given 19-base segment would occur exactly once among the patent
sequences. He should've instead calculated the likelihood that the
19-base segment would occur one or more times among the patent
sequences, which is the same as one minus the likelihood of zero
occurrences: 1-((1-.25^19)^((3300-19+1)*24712) (where
1-.25^19 is the likelihood that a segment at a given
position of a given patent sequence does not match the 19-base target
sequence, and (3300-19+1)*24712 is the number of 19-base
segments on a single strand of all patent sequences). The resulting
probability is about 0.000295014, which is only about 0.01% higher than
Ambati's P2c probability so it doesn't make much difference
though.P2c
probability by 2.I don't even know where Ambati et al. got their number of 24,712 sequences in the Moderna patent. Actually the patent contains a total of 33,915 sequences, out of which 4,573 are amino acid sequences and 406 are RNA sequences that contain U bases. The remaining number of nucleotide sequences is 28,936 which doesn't math Ambati's number of 24,712 sequences. And Ambati's guess that the median length of the sequences was about 3,300 bases was also wrong because actually the median length is 1,250.5 bases if you exclude the amino acid and RNA sequences.
But anyway, Fleming was wrong to say that "the odds of such a cleavage site occurring spontaneously (naturally) is 3.21 x 10-11". The likelihood that the FCS would occur spontaneously is not the same as the likelihood that a specific Moderna patent would happen to match the specific insert that happened to add the FCS along with 7 surrounding bases. There are also many other inserts that could've added an FCS to SARS-CoV-2 and even many combinations of point mutations that could've added an FCS. The FCS sequence only needs to have two arginine residues with two other residues in between, and the sequence can be located at multiple different spots near the S1/S2 junction.
There's 6 different codons for arginine so there's many different
ways to introduce mutations that produce an arginine residue. For
example in RaTG13 the codons after the spot where Wuhan-Hu-1 has
RRAR are AGT GTA GCT AGT which codes for
SVAS, but it would change to RVAR if the third
nucleotide was changed to A or G and the 12th
nucleotide was changed to A or G.
The codons around the S1/S2 junction in RaTG13 are
T:act N:aat S:tca R:cgt S:agt V:gtg A:gcc, which would match
RXXR if either the T at the start or the
A at the end changed to R. But there's 3
combinations of 2 nucleotide changes and 3 combinations of 3 nucleotide
changes which would change the T at the start to
R, and there's 1 combination of 2 nucleotide changes and 5
combinations of 3 nucleotide changes which would change the
A at the end to R.
The likelihood that the 12-base FCS insert would occur by chance is not even the same as the likelihood that the 12-base FCS insert and the 7 surrounding bases would occur by chance. And the likelihood that the specific FCS motif in Wuhan-Hu-1 would occur by chance is not the same as the likelihood that any FCS motif would occur by chance. And the likelihood that the FCS insert in Wuhan-Hu-1 would occur naturally is not the same as the likelihood that it would occur by chance, because certain mutations might be favored by natural selection. And the likelihood that the 19-base segment happened to match one specific Moderna patent is not the same as the likelihood that the 19-base segment would have occurred naturally.
In order to estimate the likelihood that the 12-base insert and a
total of 7 surrounding bases from either side would have an exact match
in the patent database, you can go here:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn.
Set the query sequence to ACTAATTCTCCTCGGCGGGCACGTAG, which
consists of the 12-base FCS insert, 7 bases before it, and 7 bases after
it. Then switch the database to patent sequences, insert "SARS-CoV-2 (taxid:2697049)" as the organism and
click the exclude checkbox next to it, and under algorithm parameters
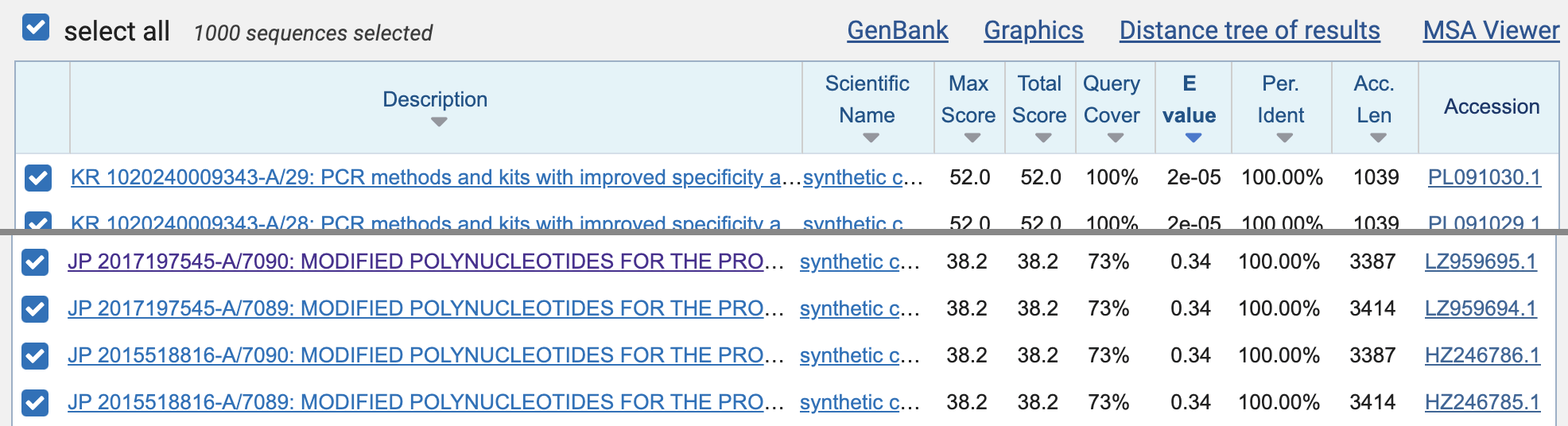
set maximum results to 1000, and then click "BLAST". When I ran the query, the first 392
matches were mostly matches to SARS-CoV-2 that were not removed by the
species filter, but the matches to the Moderna patent sequences were
listed on rank 393 to 396 (JP 2017197545-A/7090, JP 2017197545-A/7089,
JP 2015518816-A/7090, and JP 2015518816-A/7089). However the E-value of
all matches was about 0.34, which means that about 0.34 similarly close
matches are expected to occur by chance in the patent database:
The E-value depends on the size of the database, and the patent database is fairly small compared to other databases like the core nucleotide database. So when I repeated the previous search but I switched to the core nucleotide database, there were a bunch of bacterial sequences that got a perfect match to some 19-base subsegment of the 26-base query, but their E values were about 8.9 which means that about 8.9 similarly close matches were expected to occur by chance.
There's 29,885 different 19-base segments of Wuhan-Hu-1, but 1,670 or about 6% of them had an exact match to the GRCh38 human reference genome:
$ brew install bowtie2 seqkit
$ wget ftp://ftp.ccb.jhu.edu/pub/data/bowtie_indexes/GRCh38_no_alt.zip;unzip GRCh38_no_alt.zip
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947'>sars2.fa
$ seqkit seq -s sars2.fa|awk -F '' '{for(i=1;i<length-18;i++)print ">"i"\n"substr($0,i,19)}'>frag.fa
$ bowtie2 -p4 --no-unal -x GRCh38_noalt_as/GRCh38_noalt_as --score-min C,0,-1 -fU frag.fa>temp
29884 reads; of these:
29884 (100.00%) were unpaired; of these:
28214 (94.41%) aligned 0 times
1427 (4.78%) aligned exactly 1 time
243 (0.81%) aligned >1 times
5.59% overall alignment rate
Jikky's 19-base segment matches bases 23601 to 23619 of Wuhan-Hu-1,
which codes for the PRRAR codons along with 2 bases before
them and 2 bases after them. The 19-base segment matches the 12-base
insertion in Wuhan-Hu-1 and the next 7 bases. However if we go along
with Jikky's theory where SARS2 acquired the insert in a cell culture
from the human MSH3 gene, it's not clear if the 7-base flanking region
after the insert would've helped to facilitate accidental recombination
much better than a 6-base or 5-base flanking region. And if the last two
bases from Jikky's segment are left out so the flanking region is only 5
bases long, then the 17-base segment has a perfect match on BLAST to all
of these patent sequences published before 2020:
$ curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=xml&id=HC903376,HC915932,HC908933,HC905783,HB443296,GN095906,BD179519'>temp.xml $ xmlstarlet fo -D temp.xml|xml sel -t -m //GBSeq -v GBSeq_accession-version -o \| -v GBSeq_definition -o \| -v GBSeq_create-date -o \| -v .//GBReference/GBReference_title -o \| -v .//GBReference/GBReference_journal -o \| -n|awk -F\| '$3!~/202/' HC903376.1|Sequence 13528 from Patent EP2194140|18-JUN-2010|Process for the production of fine chemicals|EP2194140-A2 13528 09-JUN-2010 Metanomics GmbH (DE)| HC915932.1|Sequence 26085 from Patent EP2194140|18-JUN-2010|Process for the production of fine chemicals|EP2194140-A2 26085 09-JUN-2010 Metanomics GmbH (DE)| HC908933.1|Sequence 19086 from Patent EP2194140|18-JUN-2010|Process for the production of fine chemicals|EP2194140-A2 19086 09-JUN-2010 Metanomics GmbH (DE)| HC905783.1|Sequence 15936 from Patent EP2194140|18-JUN-2010|Process for the production of fine chemicals|EP2194140-A2 15936 09-JUN-2010 Metanomics GmbH (DE)| HB443296.1|Sequence 19 from Patent WO2009077406|14-JUL-2009|Lipid metabolism proteins, combinations of lipid metabolism proteins and uses thereof|WO2009077406-A1 19 25-JUN-2009 BASF Plant Science GmbH (DE)| GN095906.1|Sequence 687 from Patent WO2009037279|16-APR-2009|Plants with increased yield|WO2009037279-A1 687 26-MAR-2009 BASF Plant Science GmbH (DE)| BD179519.1|Highly thermophilic bacterium-derived protein and gene encoding it|15-MAY-2003|Highly thermophilic bacterium-derived protein and gene encoding it|JP2002325574-A 10 12-NOV-2002 THE INSTITUTE OF PHYSICAL AND CHEMICAL RESEARCH|
To reproduce the table above, go here:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn.
Enter CTCCTCGGCGGGCACGT to the field at the top, switch the
database to "Patent sequences(pat)", and
click BLAST. Then copy the accession numbers up to the last result where
query coverage and identity are both 100%, and run the code above with
the accession IDs joined by commas inserted as the id parameter.
The Moderna patent includes a total of 33,915 sequences. It's an unusual practice to include such a large amount of genetic material in a single patent, so McKernan called it a "kitchen sink patent". You can download all sequences from here: https://seqdata.uspto.gov/docdetail?docId=US09587003B2. Their average length is about 1,700 bases and their total length is about 58 million bases:
$ unzip US09587003-20170307-SUPP.zip $ sed 's/.*SEQUENCE: />/;/^[< ]/d;s/ *[0-9]*\r$//;s/ //g' Psips/Data/03/US/2017/870/095/B2/sequence/US09587003-20170307-S00001.TXT|seqkit stat file format type num_seqs sum_len min_len avg_len max_len - FASTA DNA 33,915 57,673,267 7 1,700.5 18,787
The Moderna patent contains 4,573 amino acid sequences and 406 RNA sequences that contain U bases. And out of the remaining 28,936 sequences, 24,363 sequences don't have any stop codons in the middle if they are translated. I believe they include all or nearly all human protein-coding genes, because the human genome contains about 20,000 protein-coding genes and their average length is about 1,500 bases:
$ wget ftp://ftp.ensembl.org/pub/release-109/fasta/homo_sapiens/cds/Homo_sapiens.GRCh38.cds.all.fa.gz $ seqkit fx2tab Homo_sapiens.GRCh38.cds.all.fa.gz|grep transcript_biotype:protein_coding|awk '!a[$7]++'|seqkit tab2fx|seqkit stat file format type num_seqs sum_len min_len avg_len max_len - FASTA DNA 19,562 29,733,131 3 1,519.9 100,272
When I generated all 19-base segments of Wuhan-Hu-1 with the poly(A) tail removed and I searched for them within the the Moderna patent, there were 3 other 19-base segments besides Jikky's segment which had a perfect match to the patent:
$ tr -d \\r<Psips/Data/03/US/2017/870/095/B2/sequence/US09587003-20170307-S00001.TXT|awk '/TYPE: DNA/' RS= 'ORS=\n\n'|sed 's/<400> SEQUENCE: />/;/^[< ]/d;s/ *[0-9]*$//;s/ //g'|seqkit grep -svp u>moderna.fa
$ bowtie2-build --thread 4 moderna.fa{,}
$ seqkit replace -sip'a*$' sars2.fa|seqkit sliding -W19 -s1|seqkit replace -p'.*:'>frag.fa
$ bowtie2 -p4 --no-unal -x moderna.fa --score-min C,0,-1 -fU frag.fa>moderna.sam
$ (echo sars2_pos patent_sequence_number match_start_in_patent_sequence sequence;grep -v ^@ moderna.sam|cut -f1,3,4,10)|column -t
sars2_pos patent_sequence_number match_start_in_patent_sequence sequence
3180-3198 18510 967 AAGAAGAGCAAGAAGAAGA
4459-4477 26445 1172 AGTTTCAACTATACAGCGT
8293-8311 30068 1160 CTATAACAAAGTTGAAAAC
23601-23619 11651 2760 CTACGTGCCCGCCGAGGAG
(The last sequence above matched both the patent sequence IDs 11651 and 11652 but Bowtie2 only returned one of the matches.)
If the amino acid and RNA sequences are excluded, the Moderna patent
has a total of about 49 million 19-base segments. And there's 29,885
19-base segments in Wuhan-Hu-1. So if both collections of segments would
be random, the expected number of exact matches between the collections
would be .25^19*49159360*29885*2 which is about 11.
Jikky wrote this about the 19-base segment: "In order for that sequence to have arisen in that virus, the virus which was manufactured with its HIV inserts, had to have had been infected into patented cell lines supplied by Moderna that had that unique sequence not seen in any other virus." [https://www.arkmedic.info/p/how-to-blast-your-way-to-the-truth] However it's possible for bioweaponeers to edit the genome as a text file and then synthesize the whole genome, so they don't have to find a cell line which contains the 12-base segment they want to insert, then infect that cell line with the virus a zillion times until the right piece of the cell line's genome happens to get inserted to the right spot of the viral genome by chance. In the comments of Jikky's Substack post, Jennifer Smith who is a PhD virologist also said that "the 'accidental' nature you implied is less likely than purposeful construction". [https://www.arkmedic.info/p/how-to-blast-your-way-to-the-truth/comment/4545048]
Jikky also stated it as a fact that SARS-CoV-2 was made using an MSH3 cell line patented by Moderna: "Irrespective of whether it was released there it distracts from the fact that it was made in a lab using a Moderna patented MSH3_mut cell line." [https://x.com/JikkyKjj/status/1474581145979293702]
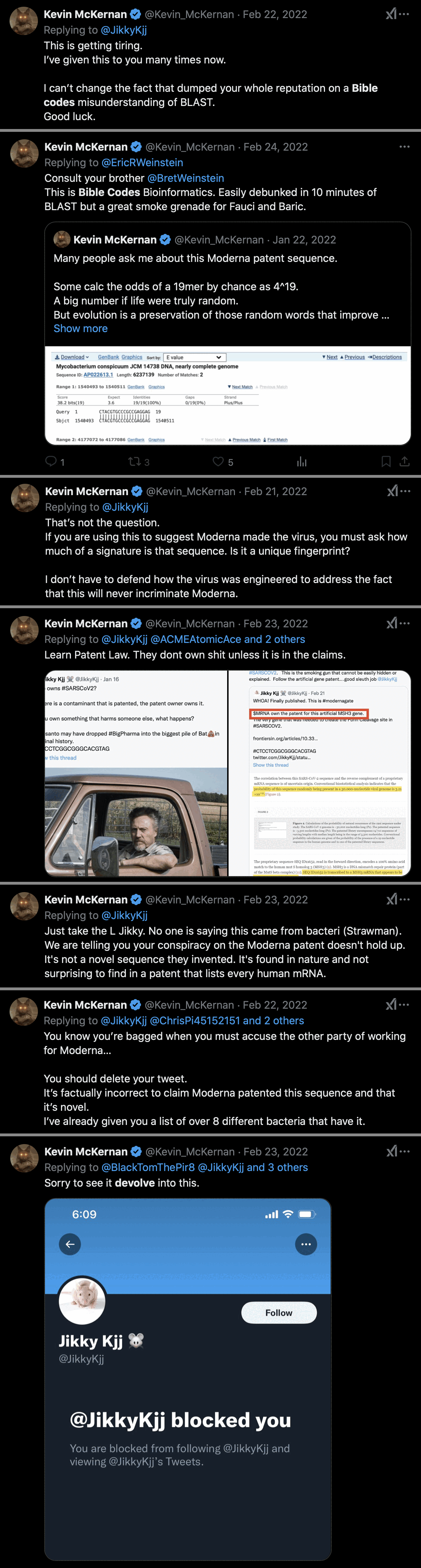
Kevin McKernan was blocked by Jikkyleaks in 2022 after McKernan debunked Jikky's Modernagate theory in this thread: https://x.com/Kevin_McKernan/status/1484987210508255233. I recommend reading the whole thread. McKernan said that Jikkyleaks had dumped his reputation on a Bible codes interpretation of BLAST: [https://x.com/search?q=from%3Akevin_mckernan+to%3Ajikkykjj+since:2022-2-10&f=live]
Added later: Modernagate was also debunked here by the Japanese patent expert Patent_SUN: https://x.com/Patent_SUN/status/1901917154578202855. For example he wrote:
The following five patent publications ① to ⑤ each includes the above reverse complementary sequence in its Sequence Listing, but no patent rights have been granted to the reverse complementary sequence itself.
①US9149506B2
②US9216205B2
③US9255129B2
④US9301993B2
⑤US9587003B2Therefore, we must carefully explain the reverse complementary sequence so as not to mislead the public. Regarding those patent publications ① to ⑤, by referring to the "We claim" section (scope of Claims) attached to the end of each specification, we can understand where the patent rights exist.
As we can see in each "We claim" section of the patent publications ① to ⑤, there is no description of "CTACGTGCCCGCCGAGGAG."
From this fact, we can understand that the patent rights have NOT been granted to it.
[...]
The topic has strayed off course, but realistically, it is not possible for Moderna to obtain a patent for this specific sequence, "CTACGTGCCCGCCGAGGAG," or just a part of it (i.e., the furin cleavage site). This is because Moderna disclosed "SEQ ID 11652" as a complete sequence, which includes that specific sequence, but did not disclose the segment as an independent invention. This fails to meet the support requirement for patentability, and it is unlikely that a patent will be granted for this specific sequence in the future.
Furthermore, even if Moderna were to attempt to obtain a patent right for "SEQ ID 11652," I believe it would be a difficult process. Realistically, if Moderna were to pursue a patent right for "SEQ ID 11652," the specification would need to explicitly describe the usage, function, and effects of "SEQ ID 11652" in order to meet the aforementioned support requirement. However, none of the specifications in the Patents 1 to 5 mention "SEQ ID 11652," nor is there any disclosure suggesting it.
Additionally, Moderna does not address the complementary sequence of "CTACGTGCCCGCCGAGGAG," which is "CTCCTCGGCGGGCACGTAG," anywhere in the specification. Therefore, the patent rights of the Patents 1 to 5 never extend to the 'CTCCTCGGCGGGCACGTAG' sequence of the COVID-19 virus.
Another addendum: I went to nucleotide BLAST:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn.
I entered the accession of SARS-CoV-2 to the text field at the top
(NC_045512), and I changed the database to patent sequences. I set the
Entrez query to 0:2019[dp] Moderna to return patent
sequences that were published at GenBank before 2020 and that matched
the word Moderna in any of the fields. Then under algorithm parameters I
reduced the word size to 16, I increased the expect threshold to 1000,
and I increased maximum target sequences to 1000.
There were a total of 495 results. The best matches with 100% identity were 23 bases long, but even they had an expect value of about 2.3, which means that about 2.3 similarly close matches were expected to occur by chance:
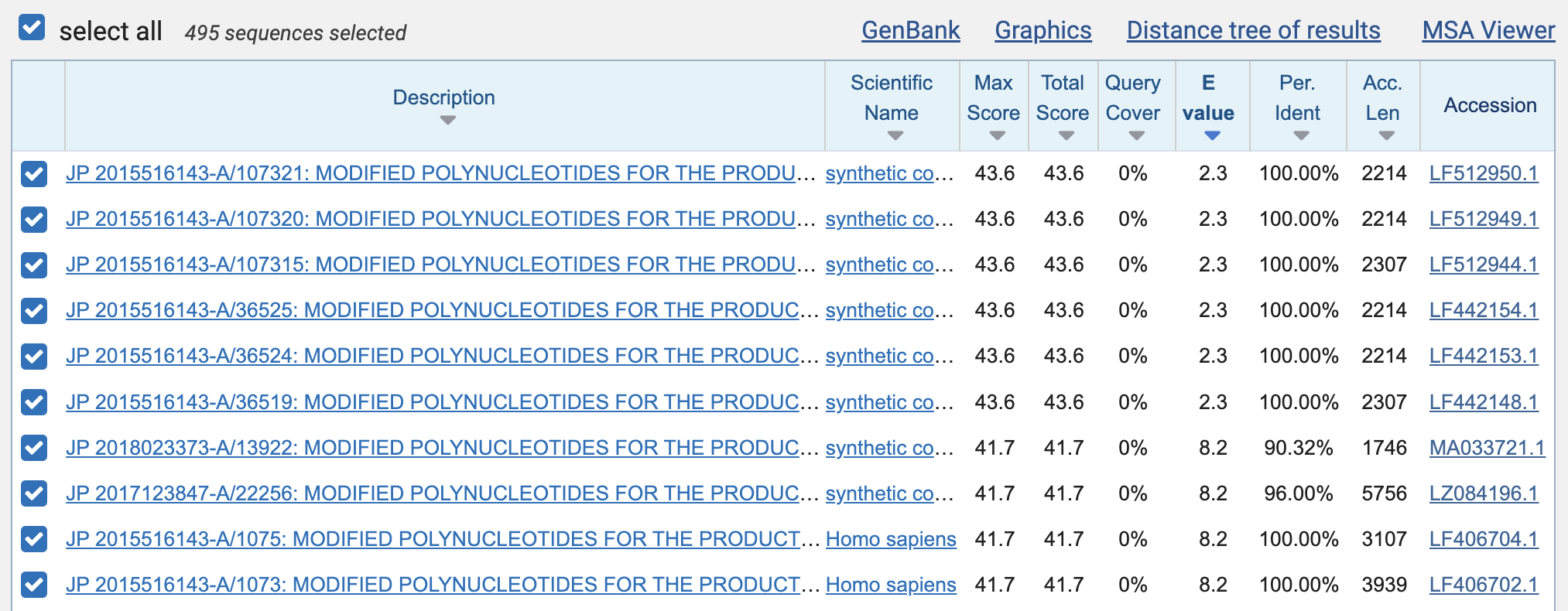
The expect values of the perfect 19-base matches were about 381, which means that about 381 similarly close matches were expected to occur by chance within the set of patent sequences that were published before 2020 and that matched the word "Moderna".
Then I clicked "Download" and selected "Hit Table (text)". It showed that there were 176 perfect matches with a length of 19 bases, 55 with length 20, 6 with length 21, 13 with length 22, and 6 with length 23:
$ awk '$3==100&&$4>=19{a[$4]++}END{for(i in a)print i,a[i]}' Downloads/XRM91BX9013-Alignment.txt
19 176
20 55
21 6
22 13
23 6
There weren't even that many duplicate matches among the perfect matches that were at least 19 bases long, because the matches started at 92 unique positions of the genome of SARS-CoV-2. There were also 256 unique patent sequences which had a perfect match at least 19 bases long:
$ awk '$3==100&&$4>=19' Downloads/XRM91BX9013-Alignment.txt|cut -f7|sort -u|wc -l 92 $ awk '$3==100&&$4>=19' Downloads/XRM91BX9013-Alignment.txt|cut -f2|sort -u|wc -l 256
The perfect matches that were at least 19 bases long covered 1,532 bases of the genome of SARS-CoV-2, which is about 5% of the total length of the genome:
$ awk '$3==100&&$4>=19{for(i=$7;i<=$8;i++)a[i]}END{print length(a)}' Downloads/XRN2HBFD016-Alignment.txt
1532
When I clicked "Search Summary", it showed
that there were a total of about 700,000 sequences in the patent
database that matched the Entrez query 0:2019[dp] Moderna,
and their total length was about a billion bases:
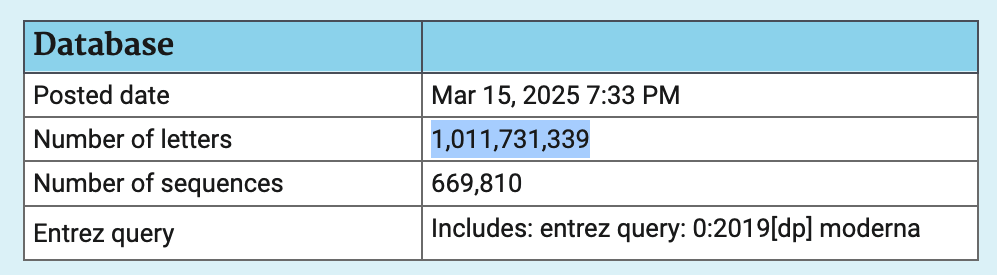
The total length of all sequences in the patent database in March 2025 was about 28.3 billion bases. So about 4% of the total length was made up by sequences that were published before 2020 and that matched the keyword "Moderna".
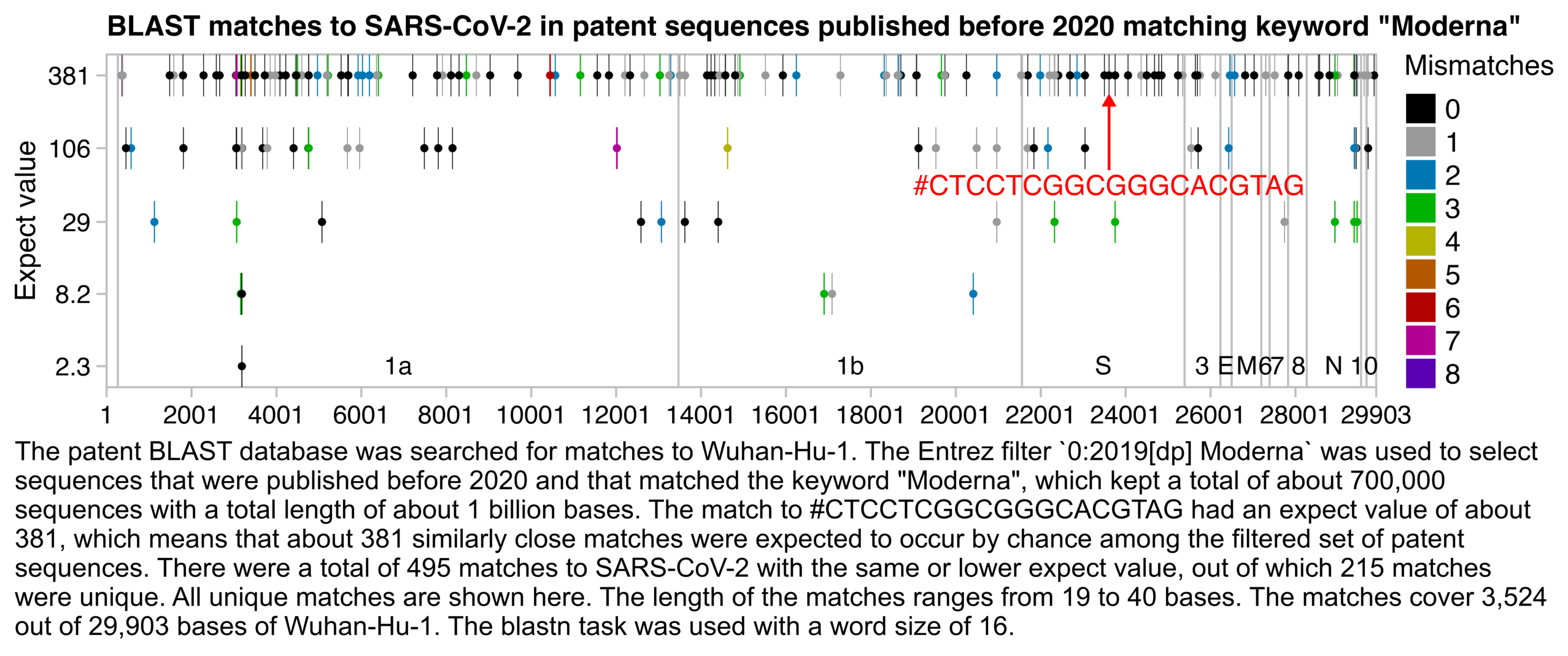
This plot shows all matches with the same or lower expect value as the match to Jikky's 19-base segment:
t=fread("http://sars2.net/f/modernagate-blast.txt")
p=unique(t[,.(start=qstart,end=qend,expect=evalue,mismatch=mismatch+gaps)])
gene=fread(text="name,start,end
1a,266,13468
1b,13468,21555
S,21563,25384
3,25393,26220
E,26245,26472
M,26523,27191
6,27202,27387
7,27394,27759
8,27894,28259
N,28274,29533
10,29558,29674")
bars=gene[,c(start[1],pmean(start[-1],end[-.N]),end[.N])]
exp=.16;ylim=10^(c(-1,1)*exp+log10(range(p$expect)))
ybreak=unique(p$expect);xend=29903;xbreak=c(seq(1,xend,2e3),xend)
color=c("black","gray60",hsv(c(20,12,6,3,0,31,27)/36,1,.7))
annox=23601+10;annoy=10^(log10(381)-exp);annoy2=10^mean(log10(ybreak[3:4]))
ggplot(p)+
annotate("rect",xmin=1,xmax=xend,ymin=ylim[1],ymax=ylim[2],linewidth=.4,lineend="square",color="gray75",fill=NA)+
geom_vline(xintercept=bars,color="gray75",linewidth=.4,lineend="square")+
geom_rect(aes(fill=factor(mismatch),xmin=start,xmax=end,ymin=10^(log10(expect)-exp),ymax=10^(log10(expect)+exp)))+
geom_point(aes(color=factor(mismatch),x=pmean(start,end),y=expect),size=1.5,stroke=0)+
geom_text(data=gene,aes(x=pmean(start,end),y=ylim[1],label=name),size=3.5,hjust=.5,vjust=-.7)+
annotate("segment",x=annox,xend=annox,y=annoy2*1.3,yend=annoy,arrow=arrow(type="closed",length=unit(4,"pt")),lineend="butt",linejoin="mitre",color="red",size=.5)+
annotate("text",x=annox,y=annoy2,size=3.8,color="red",label="#CTCCTCGGCGGGCACGTAG")+
labs(x=NULL,y="Expect value",title="BLAST matches to SARS-CoV-2 in patent sequences published before 2020 matching keyword \"Moderna\"")+
scale_x_continuous(limits=c(1,xend),breaks=xbreak)+
scale_y_log10(breaks=ybreak,labels=ifelse(ybreak>=10,round(ybreak),ybreak))+
scale_color_manual(values=color,name="Mismatches")+
scale_fill_manual(values=color,name="Mismatches")+
coord_cartesian(clip="off",expand=F)+
theme(axis.text=element_text(size=10,color="black"),
axis.text.x=element_text(margin=margin(3)),
axis.ticks=element_line(size=.4,color="gray75"),
axis.ticks.length=unit(4,"pt"),
legend.background=element_blank(),
legend.key=element_blank(),
legend.key.height=unit(13,"pt"),
legend.key.width=unit(13,"pt"),
legend.spacing.x=unit(3,"pt"),
legend.text=element_text(size=11),
panel.background=element_blank(),
plot.margin=margin(5,5,5,5,"pt"),
plot.title=element_text(size=11,face=2,margin=margin(1,,5)))
ggsave("1.png",width=8.5,height=2.4,dpi=300*4)
sub="The patent BLAST database was searched for matches to Wuhan-Hu-1. The Entrez filter `0:2019[dp] Moderna` was used to select sequences that were published before 2020 and that matched the keyword \"Moderna\", which kept a total of about 700,000 sequences with a total length of about 1 billion bases. The match to #CTCCTCGGCGGGCACGTAG had an expect value of about 381, which means that about 381 similarly close matches were expected to occur by chance among the filtered set of patent sequences. There were a total of 495 matches to SARS-CoV-2 with the same or lower expect value, out of which 215 matches were unique. All unique matches are shown here. The length of the matches ranges from 19 to 40 bases. The matches cover 3,524 out of 29,903 bases of Wuhan-Hu-1. The blastn task was used with a word size of 16."
system(paste0("mogrify -trim 1.png;magick 1.png \\( -size `identify -format %w 1.png`x -font Arial -interline-spacing -3 -pointsize $[43*4] caption:'",gsub("'","'\\\\'",sub),"' -splice x80 \\) -append -resize 25% -bordercolor white -border 24 -dither none -colors 256 1.png;pngcrush -rem alla -reduce 1.png 1.png"))
When I searched the nucleotide database at GenBank for the query
0:2019[dp] moderna patent, there were a total of 671,931
results, which is close to the same as the number of sequences in the
patent BLAST database that matched the query
0:2019[dp] moderna.
[https://www.ncbi.nlm.nih.gov/nuccore/?term=0%3A2019%5Bdp%5D+moderna+patent]
Fleming's affidavit referred to Alex Washburne's paper titled "Endonuclease fingerprint indicates a synthetic origin of SARS-CoV-2".
The paper said: "Fisher's exact test was used to assess if there was a higher rate of silent mutations within BsaI/BsmBI recognition sites compared to the rest of the viral genome. Odds ratios were computed as the ratio of silent mutations to all other nucleotides within BsaI/BsmBI sites of either genome in a pairwise alignment compared to the ratio of silent mutations outside BsaI/BsmBI sites to all other nucleotides. There are 12 silent mutations found in 9 distinct BsaI/BsmBI sites between RaTG13 and SARS-CoV-2, and 882 silent mutations outside of BsaI/BsmBI sites." [https://europepmc.org/article/ppr/ppr560730]
The BsaI recognition sequence is GGTCTC and the BsmBI
recognition sequence is CGTCTC, so both of them are 6 bases
long. So the total length of the 9 recognition sites is 9*6
which is 54 bases.
So the Fisher's test can be calculated like this:
recognition_silent=12 recognition_total=54 other_silent=882 genome_length=29903 m=c(recognition_silent,recognition_total-recognition_silent) m=c(m,other_silent,genome_length-other_silent-recognition_total) fisher.test(matrix(m,2))$p # 5.120702e-08
However Guy Gadboit showed here why Washburne's logic of calculating the p-values and odds ratios was wrong: https://x.com/gadboit/status/1833191562961887679. After Gadboit corrected the calculation, he got the odds ratios to be centered around 1 and not around 2 like in Washburne's code. See also these threads: https://x.com/gadboit/status/1748538944717750619, https://x.com/gadboit/status/1702271854671462655.
There's 5 BsaI/BsmBI recognition sites in Wuhan-Hu-1 and 6 in RaTG13, but 2 of them are included in both so there's 9 distinct sites:
$ curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947'>sars2.fa $ seqkit locate -rip'ggtctc|cgtctc' sars2.fa|cut -f4-5,7|column -t strand start matched - 24102 GGTCTC - 17972 GGTCTC # included in both - 17329 CGTCTC # included in both - 9751 CGTCTC - 2193 CGTCTC $ seqkit locate -rip'ggtctc|cgtctc' ratg13.fa|cut -f4-5,7|column -t strand start matched + 11645 ggtctc + 22919 cgtctc + 24494 cgtctc - 17969 ggtctc # included in both - 17326 cgtctc # included in both - 10441 ggtctc
People often say that the BsmBI and BsaI sites are ideally spaced for a genetically engineered virus. But the output above shows that the two sites that are shared by both RaTG13 and Wuhan-Hu-1 start at positions 17326 and 17969 in RaTG13. So why would two such nearby sites be kept? But on the other hand the site 24494 would have been removed and 24102 would have been inserted in its place. And 10441 would be removed but 9751 would be inserted in its place.
In the scenario where a restriction map was built for Wuhan-Hu-1 using RaTG13 as a starting point, the engineers of the virus might have introduced silent mutations to eliminate unwanted recognition sites and to introduce wanted recognition sites, which is why Washburne counted the number of recognition sites in both the source and target sequence. His paper said: "In 2013, researchers constructed a recombinant MERS coronavirus (Scobey et al. 2013). The wildtype virus had a few BglI sites at inconvenient locations, making it poorly amenable to efficient assembly. To construct an idealized MERS-CoV reverse genetic system for IVGA, the researchers removed the existing BglI sites and inserted 6 more evenly spaced BglI sites. All additions/removals were done via synonymous mutations, creating 7 fragments, the longest of which was 5721bp, or 19% the length of the 30kb MERS genome (Fig 2A)."
I don't know why the engineers would need to use both BsaI and BsmBI and not just one of them, like in the case of the reverse genetics system for MERS where only BglI was used. The draft version of the DEFUSE proposal mentioned an order for BsmBI (R0580S and R0580L) but not BsaI: [https://x.com/NestCommander/status/1783112388000256308]

If only BsmBI was used, it would produce only 4 fragments where the longest fragment would be about 12,000 bases long:
$ seqkit locate -rip'cgtctc' sars2.fa|cut -f4-5,7|column -t strand start matched - 17329 CGTCTC - 9751 CGTCTC - 2193 CGTCTC
Washburne's paper says that "the BsaI sites in SARS-COV-2 flank the S1 gene and S1/S2 junction, and a similar design has been used before for substitutions in this region". However one BsaI site is about 500 away from the S1/S2 junction and the other site is about 3600 bases away from the start of S1:
$ seqkit locate -rip'ggtctc,...cggcgg...' sars2.fa|cut -f4-5,7|column -t strand start matched + 23603 CCTCGGCGGGCA # FCS - 24102 GGTCTC # BsaI site (about 500 bases away from S1/S2 junction) - 17972 GGTCTC # BsaI site (about 3600 bases away from start of S1)
Restriction enzymes are not even needed to make synthetic coronaviruses. Stuart Neil wrote: "With all this talk about restriction enzyme smoking guns, perhaps it's time to admit (oh the shame) that my lab can make coronavirus genomes and mutants thereof without using yeast, bacteria or restriction enzymes. And we can do it over night and rescue the virus in a week." [https://x.com/stuartjdneil/status/1748367338590593116] And another Twitter user wrote: "I hope I'm not being irresponsible when I say that restriction enzymes have been passe for at least a decade now. We reverse engineer viruses weekly using Gibson assembly (fancy PCR). Restriction sites? Pish tosh!!!" [https://x.com/ruby_marchant/status/1748452331887452635] And Billy Bostickson wrote: "Cloning of full genome cDNA in a plasmid is bypassed using the ISA method relying on ex vivo recombination & transcription of viral RNA. Thus, no restriction sites or other genomic modifications are required. https://embopress.org/doi/full/10.15252/embr.202153820" https://x.com/BillyBostickson/status/1748632049010053569]
Fleming made this image which showed that Corman-Drosten's RdRp primers matched MA15 and the SARS1-like bat viruses SHC014 and Rs3367: [https://www.flemingmethod.com/gain-of-function]

Corman-Drosten's RdRp primer set was designed to match both SARS-CoV-2 and other sarbecoviruses including SARS-CoV. It has two different probes, where the P2 probe is designed to be specific to SARS-CoV-2 but the P1 probe is a so-called "pan-sarbecovirus" probe that is also designed to match other SARS-like viruses. So it's no surprise that the RdRp primers actually match the MA15 strain of SARS1 and SHC014 and Rs3367 which are SARS1-like bat viruses, because that's what they were designed to do.
The paper about the Corman-Drosten protocol includes the figure below which shows that their primers match various SARS-like viruses like ZC45, SARS1, and BM48-31 (even though the E gene primers and probe each have a single mismatch to BM48-31, and the N gene primers and probe have even more mismatches to BM48-31, so the N gene primers and probe might have too many mismatches to yield a positive result for BM48-31): [https://www.eurosurveillance.org/content/10.2807/1560-7917.ES.2020.25.3.2000045]

I also don't understand why Fleming claims that MA15 is a "chimera", because a chimeric virus has to be a combination of two or more viruses. The paper about MA15 said: "We adapted the SARS-CoV (Urbani strain) by serial passage in the respiratory tract of young BALB/c mice. Fifteen passages resulted in a virus (MA15) that is lethal for mice following intranasal inoculation." [https://pubmed.ncbi.nlm.nih.gov/17222058/] There's a large number of different MA15 sequences at GenBank, but Fleming's screenshot showed the MA15 isolate d30m5, which has only 23 nucleotide changes from the SARS-CoV Urbani sequence at GenBank:
$ curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=AY278741.1,JF292920.1'|mafft --thread 4 --quiet ->urbanima15.fa
$ seqkit seq -s urbanima15.fa|awk 'NR==1{split($0,a,"");next}{split($0,b,"");d=0;for(i=1;i<=length;i++)d+=a[i]!="-"&&b[i]!="-"&&a[i]!=b[i];print d}'
23
SHC014 and Rs3367 are also not publicly known as either chimeric viruses or gain-of-function viruses (even though the many recombination events around the NTD and RBM in SARS1-like viruses are of course suspicious, and it's also strange how Rs3367 is otherwise nearly identical to WIV1 but Rs3367 is missing ORFx but WIV1 is not). Fleming may have confused SHC014 with the chimeric virus described in Menachery et al. 2015, where the spike protein of SHC014 was inserted to an MA15 backbone. In another version of his graphic I showed above he has in fact inserted the SHC014-MA15 chimera in place of the regular SHC014. However even in the SHC014-MA15 chimera the RdRp comes from SARS1, so it's not unusual that the RdRp would match Corman-Drosten's primers which were designed to match SARS1.
SHC014 and Rs3367 both have about 95-96% nucleotide identity to SARS1. And in both of them the region of the RdRp that was shown in Fleming's image is 100% identical to the Tor2 reference genome of SARS1:
$ curl -sL sars2.net/f/sarbe.fa.xz|xz -dc>sarbe.fa $ seqkit grep -nrpTor2,RsSHC014,Rs3367 sarbe.fa|seqkit seq -g|seqkit locate -ip attaagtgagatggtcatgtgtggcggctcactatatgttaaaccaggtggaacatcatccggtgatgctacaactgcttatgctaatagtgtctttaacatttgtcaag|cut -f1,4-6|column -t seqID strand start end KC881006.1 + 15356 15465 KC881005.1 + 15356 15465 NC_004718.3 + 15356 15465
In my FASTA file of 529 sarbecovirus sequences, Corman-Drosten's RdRp forward primer has an exact match to 482 sequences:
$ seqkit seq -g sarbe.fa|seqkit stat file format type num_seqs sum_len min_len avg_len max_len - FASTA DNA 529 15,673,697 25,965 29,628.9 30,600 $ seqkit seq -g sarbe.fa|seqkit locate -dip GTGARATGGTCATGTGTGGCGG|sed 1d|wc -l 482
And the RdRp reverse primer has an exact match to 444 out of 529 sequences in my file:
$ seqkit seq -g sarbe.fa|seqkit locate -dip CARATGTTAAASACACTATTAGCATA|sed 1d|wc -l 444
Fleming wrote:
By Doing This You Will Find That SARS-CoV-2 Wuhan Hu-1 Matches Three Gain-of-Function (GoF) Viruses Built by Baric and Zhengli Using NIAID and Other Federal Money.
The Three GoF Viruses are:
CoV-RsSHC014
(aka. SARS-CoV-Urbani 2003 & 2016)
SARS-CoV-MA15
SARS-CoV-Rs3367
Fleming also made an image which said: "Matching PCR Primers for SARS-CoV-2 Wuhan-Hu-1 Using Snap Gene Analysis Reveal Matches with SARS-CoV-Rs3367, CoV-RsSHC014 (Urbani) & SARS-CoV-MA15." [https://x.com/Doctor_I_am_The/status/1886410702937641471] I believe he suggested that SHC014 was the same virus as SARS-CoV Urbani. He may have been confused by how at GenBank Baric's MA15-SHC014 chimera is called "SARS-Urbani-MA_SHC014-spike". [https://www.ncbi.nlm.nih.gov/nuccore/MT308984.1] But MA15 is a mouse-adapted strain of SARS-CoV which is derived from the Urbani strain, and in Baric's chimera the spike protein of the SARS1-like bat virus SHC014 was inserted to MA15.
I also don't understand what evidence Fleming has that specifically Rs3367 was "built by Baric and Zhengli" like he wrote. It's one of about 50 published SARS1-like bat viruses that have around 5-6% nucleotide distance to SARS1. I do consider some of the SARS1-like bat viruses to have a laboratory origin, and it's suspicious how Rs3367 is otherwise nearly identical to WIV1 except Rs3367 is missing ORFx but WIV1 is not, and there are similarly other branches of SARS1-like viruses where ORFx is included in some members of the branch but not others, which is reminiscent of how an ACE2-binding RBM seems to have mysteriously emerged in some closely related SARS1-like viruses but not others. [https://academic.oup.com/view-large/figure/232195290/veab007f2.tif] But I haven't seen Fleming explain why he has specifically focused on Rs3367.
Fleming also wrote:
All of which raises an interesting question.
We have been thinking about SARS- CoV-2 as a single virus because we have been told it's a single virus; but
What if what we have been calling SARS-CoV-2 isn't a single Gain-of-Function (GoF) Corona Virus leaked out of the Wuhan Lab?
What if it's MORE THAN ONE? Why should we believe that only one GoF virus leaked?
However if other sarbecoviruses would've been in widespread circulation then they would've been detected by metagenomic sequencing, where all genetic material in a sample is sequenced without having to rely on primers to amplify any targeted organism: nopandemic.html#Was_SARS_CoV_2_circulating_in_humans_before_2020_but_not_detected_in_the_same_way_that_HKU1_and_NL63_were_only_discovered_after_SARS1. The NCBI provides cloud services that make it possible to search the Sequence Read Archive for sequencing runs with reads that have a k-mer hit for a specific virus like SARS-CoV. A lot of people including me have searched through sequencing runs trying to look for unusual SARS-like viruses, and I have downloaded thousands of SRA runs with k-mer hits for sarbecoviruse and I have aligned the reads of the runs against SARS-like viruses, but I haven't found evidence that any other sarbecovirus besides SARS-CoV-2 would've circulated in humans since 2020. In 2021 Chinese researchers found French influenza sequencing runs from 2007-2012 that were contaminated with the lab-created ExoN1 and wtic strains of SARS1 (so the runs were probably either contaminated in a laboratory or there was an outbreak of SARS1 in France due a laboratory leak). [https://www.sciencedirect.com/science/article/pii/S2590053621001075] But if SARS1-like viruses would've been in any kind of widespread circulation since 2020, then people would've found reads of SARS1 at the SRA by using similar methodology that the Chinese researchers used to find the French influenza A samples that were contaminated with SARS1. And in general a huge amount of resources was directed to researching sarbecoviruses during COVID, so if other sarbecoviruses woudl've been in any kind of widespread circulation in humans, they would have certainly been detected.
Fleming also posted a tweet that said: "People did not die because the PCR test wasn't conducted according the the patent. They weren't maimed because of failure to conduct the PCR test pursuant to the patent. They died because of SARS-CoV-Rs3367, CoV-RsSHC014 and SARS-CoV-MA15." [https://x.com/Doctor_I_am_The/status/1843334021629030776] But I don't know how he was confident enough that specifically Rs3367, SHC014, and MA15 were circulating in humans that he was willing to state it as a fact that people died because of those specific viruses. Apart from SHC014 and Rs3367, there are approximately 50 other SARS1-like viruses published at GenBank that have around 95% identity to SARS1, so how did Fleming know it wasn't one of them circulating in humans instead?
Fleming said: "In 2001, Ralph Baric at the University of North Carolina gets a patent. It was paid for by National Institutes of Health Research. And its specific goal was organizing chromosomes of viruses. He liked transmissible gastroenteritis virus at the time - it was what he was working on - infectious bronchitis, and coronaviruses." [https://www.flemingmethod.com/gain-of-function, presentation in Phoenix, time 15:27]
However viruses obviously don't have chromosomes (even though the genomes of segmented viruses like influenza viruses are split into multiple segments akin to chromosomes in eukaryotes and prokaryotes). Fleming might have been confused by the title of the patent which was "Directional assembly of large viral genomes and chromosomes", even though it obviously meant "large viral genomes" and "chromosomes of other organisms". [https://patents.google.com/patent/US4683195A/en]
The authors of the patent wrote that their method of assembly can also be applied to chromosomes of bacteria, plants, and animals: "The genomes of viruses, bacteria, plants and other organisms (including humans) are being systematically cloned and sequenced. Methods are needed to directionally assemble smaller DNA subclones into full-length, functionally intact genomes or chromosomes of these organisms. Such methods could allow for the precise genetic manipulation of individual chromosomes in whole plants and animals and the construction of artificial chromosomes for gene therapy."
Fleming said: "But once you know you have a new
virus, what do you do? Well you have to get cell tissue out, like those
bronchoalveolar lavages. You clean up the garbage, you get rid of the
other material, you get rid of the bacteria, all the mucus, all the
garbage that's in there, and you're left with genetic code
sequences."
[https://www.flemingmethod.com/gain-of-function,
part 2 of presentation in Tampa, time 11:09] However in the paper by Wu
et al. which described the first published genome of SARS-CoV-2, the
authors did metagenomic sequencing where they sequenced all genetic
material in a sample of bronchoalveolar fluid but they didn't remove
human or bacterial genetic material. In their STAT results only about
0.2% of their reads are identified as viral.
[https://trace.ncbi.nlm.nih.gov/Traces/?view=run_browser&page_size=10&acc=SRR10971381&display=analysis]
About half of their reads consist of only N bases because
human reads were masked with N bases for privacy reasons.
Supplementary Table 1 of the paper shows that 49 out of 50 of the most
abundant MEGAHIT contigs consisted of fragments of the genomes of
bacteria.
[https://www.nature.com/articles/s41586-020-2008-3#Sec13]
Next Fleming said: "Now frequently it's broken into pieces and you have to put it together like a puzzle. But you can do that just like a puzzle, and that's called Sanger sequencing." But the procedure of assembling the reads like a puzzle is not called Sanger sequencing but it's called genome assembly. Wu et al. used next-generation sequencing and not Sanger sequencing. Sanger sequencing is used only rarely to sequence SARS-CoV-2. The NCBI's Sequence Read Archive has raw reads of about 7 million sequencing runs where the organism is listed as SARS-CoV-2, but I haven't found a single SRA run where Sanger sequencing was used to sequence the entire genome, but I have only found runs where fragments of the virus like a part of the spike protein were sequenced with Sanger sequencing, and Sanger sequencing is sometimes also used to sequence amplicons produced by PCR tests. [https://www.ncbi.nlm.nih.gov/sra/?term=sars-cov-2+%22sanger+sequencing%22]
Next Fleming described PCR testing like this: "And so the spike protein was chosen. And you already saw that we know where the spike protein is in this sequence. And so you look for the beginning of it, and you find about 12 to 18 pieces - amino acids - rather nucleotide bases - that start the spike protein, and that's called the forward primer. And you look for the end of it and you say, well, where's the end? And you look for a roughly 15 to 18 of those, and that's called a reverse primer, which reads the opposite direction." But the spike protein of Wuhan-Hu-1 is 3,822 bases long if you include the stop codon, but typically in PCR tests for SARS-CoV-2 the distance between the forward and reverse primers is only about 100 to 200 bases. The forward and reverse primers don't have to be located at opposite ends of the same gene or even within the same gene. Also PCR primers are almost always longer than 12 bases. In PCR tests for SARS-CoV-2, the length of the primers ranges from about 18 to 26 bases.
This section deals with Fleming's book "Is COVID-19 a Bioweapon?: A Scientific and Forensic Investigation". [http://libgen.li/index.php?req=richard+fleming+bioweapon]
Fleming wrote:
In 2006, using chimeric (Gain-of-Function) research, Chinese scientists reported their ability to combine parts of four different viruses into a single viral genome.[16] This report raises a few serious questions in my mind.
First, why were these researchers combining parts of four dangerous viruses - specifically, hepatitis C virus (HCV), human immunodeficiency virus -1 (HIV-1), SARS-CoV-1 (identified as SARS-CoV-1 and not SARSCoV), and SARS-CoV-2?
Second, if as we've been told, SARS-CoV-2 didn't appear until 2019 and there were no identified naturally occurring SARS-CoV-2 reported between this 2006 publication and 2019, then doesn't this at least in part suggest that SARS-CoV-2 is not naturally occurring but man-made?
Third, if the answer to question number two is that the virus is manmade, going as far back as 2006, then doesn't this add credence to those who have cautioned that SARS-CoV-1 was a bioweapon and SARS-CoV-2 is an upgraded version of that bioweapon?
However in the paper he referred to, the authors used "SARS-CoV1" and "SARS-CoV2" as names of two segments of SARS-CoV they inserted into a 1,200-base chimeric RNA sequence. The paper said: "Because most commercial armored RNA preparations contain exogenous sequences of <500 nucleotides, separate armored RNA species are often prepared for calibration of each target in multiple virus assays. To reduce costs and simplify multivirus detection, we are seeking to produce a single chimeric armored RNA species that might be used as a positive control for multiple viral targets. We consider this task to be feasible because the inventors of armored RNA predicted that, theoretically, at least 2 kb of nonbacteriophage RNA sequence might be encapsulated. As proof of this principle, we tried to directly package a 1200-nucleotide-long foreign RNA sequence containing gene fragments of hepatitis C virus (HCV), HIV-1, severe acute respiratory syndrome coronavirus 1 (SARS-CoV1), and SARS-CoV2 into the original armored RNA production vector pAR-1." [https://pmc.ncbi.nlm.nih.gov/articles/PMC7542541/]
The full chimeric sequence is shown in the supplementary document file:

The formatting of the document was screwed up, but text at the top says that the segment labeled "SARS-CoV2" was 190 bases long, so you can tell it refers to the segment that was highlighted in gray near the end. It matches bases 18,153 to 18,342 of the Tor2 reference genome of SARS-CoV:
$ curl -s 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=NC_004718.3'>sars1.fa $ seqkit locate -pATGAATTACCAAGTCAATGGTTACCCTAATATGTTTATCACCCGCGAAGAAGCTATTCGTCACGTTCGTGCGTGGATTGGCTTTGATGTAGAGGGCTGTCATGCAACTAGAGATGCTGTGGGTACTAACCTACCTCTCCAGCTAGGATTTTCTACAGGTGTTAACTTAGTAGCTGTACCGACTGGTTATG sars1.fa|cut -f1,4-6|column -t seqID strand start end NC_004718.3 + 18153 18342
Fleming later referred to the same paper again here: "While most people believe that SARS-CoV-2 first appeared in 2019, evidence shows the virus responsible for the InflammoThrombotic disease known as COVID-19 was being manipulated two provinces from Wuhan in 2006, and the work continued forward.[34] Those initial genetic sequences are shown in the appendix." However I guess he didn't actually bother checking if the genetic sequence in the appendix matched SARS-CoV-2 or the original SARS-CoV.
There's also another paper from 2007 where the authors constructed a 2,248-base chimeric RNA sequence which included three different segments from SARS-CoV, which they referred to as "SARS-CoV1", "SARS-CoV2", and "SARS-CoV3". The paper said: "An exogenous chimeric sequence 2,248 bp in length comprising the following sequences was inserted into a pACYCDuet-1 plasmid (p15A-type replication origin; Novagen): M-300 (nt 17∼373, 357 bp from avian influenza virus matrix gene; GenBank accession no. DQ864720), SARS-CoV1 (nt 15224 to 15618, 395 bp from SARS-CoV; GenBank accession no. AY864806), SARS-CoV2 (nt 18038 to 18340, 303 bp from SARS-CoV; GenBank accession no. AY864806), SARS-CoV3 (nt 328110 [sic; should be nt 28110] to 28692, 583 bp from SARS-CoV; GenBank accession no. AY864806), a pac site (19 bp), HCV (nt 18 to 310, 293 bp from HCV 5'UTR; GenBank accession no. AF139594), and HA300 (nt 295 to 611, 317 bp from H5N1 avian influenza virus; GenBank accession no. DQ864720)." [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2395109/] The three segments of SARS-CoV were taken from a sequence for the SARS-CoV isolate BJ202 that was submitted to GenBank in 2004. [https://www.ncbi.nlm.nih.gov/nuccore/AY864806]
Fleming wrote: "During the summer of 2019, the Wuhan Institute of Virology genetic databank records, including its viral genomes and research, were wiped - months before the recognition of the emergence of SARS-CoV-2."
However DRASTIC's paper about the database said that the website of the database became inaccessible on September 12th 2019 in an unspecified timezone (which would usually be considered fall and not summer). And it doesn't mean that the entire database would've been wiped, since later the website became intermittently accessible again: "Batvirus.whiov.ac.cn had been online for a few years, saw a version 2 released in June 2019, went inactive for a week during the second half of August 19, before becoming definitely inaccessible (out of the WIV at least) on the 12th Sep 19. It was online intermittently after this date from mid-December 2019, and occasionally until February 2020, but was not accessed from outside of the WIV after 12 September 2019." [https://www.researchgate.net/publication/349073738_An_investigation_into_the_WIV_databases_that_were_taken_offline]
Fleming wrote:
Patent number 4,683,195 was granted to Mullis and others for "a process for detecting the presence or absence of at least one specific nucleic acid sequence in a sample containing a nucleic acid or mixture of nucleic acids."
A clear review of this patent shows that Mullis did not exceed fifteen to twenty cycles of PCR for the identification of genetic material.
However the table appears to simply demonstrate how the number of copies produced by PCR doubles each cycle, so it's not an indication of how many PCR cycles Mullis used himself during various workflows.
Right before the table Fleming showed, the patent by Mullis said "If the sample is human genomic DNA, preferably the number of cycles is from about 10-30." [https://patents.google.com/patent/US4683195A/en]
A paper from 1988 where Mullis was the corresponding author said they used 60 cycles during one workflow: "Fifteen 1-μg samples of the 10-6 dilution of MOLT4 DNA in GM2064 DNA prepared previously (Fig. 2) were amplified for 60 cycles with 55°C annealing, and the primers RS79 and RS80." [http://sci-hub.ee/https://www.science.org/doi/10.1126/science.2448875, https://x.com/TakethatCt/status/1793390427594240368] And another part of the paper said: "Samples of human genomic DNA were subjected to 20 to 35 cycles of PCR ampification with optimal amounts of either Klenow or Tag DNA polymerase and analyzed by agarose gel electrophoresis (Fig. 1A) and Southern hybridization (Fig. 1B)."
Fleming wrote:
In 2010, Shi Zhengli conducted chimeric research on SARS-CoV-1 (then called SARS-CoV), including combining HIV-pseudovirus to look at the binding capacity of this virus with human ACE2 receptors. Their work specifically included altering (through mutagenesis) the spike proteins to determine how to increase the spike protein binding to the ACE2 receptor.
The research showed no proline-arginine-arginine-alanine (PRRA) insert critical to the binding of the SARS-CoV-2 to ACE2 receptors on human cells. As noted in this published research (jointly done by Dr. Zhengli at the Wuhan Institute of Virology, researchers in Australia, and the University of Minnesota Medical School), not only is the spike protein from horseshoe bats unable to bind to ACE2 receptors, but also these differences along with differences in civets highlight a critical missing piece to the zoonotic theory of the original of SARS-CoV-2:
However, although the genetically related SARS-like coronavirus (SL-CoV) has been identified in horseshoe bats of the genus Rhinolophus [5, 8, 12, 18], its spike protein was not able to use the human ACE2 (hACE2) protein as a receptor. Close examination of the crystal structure of human SARS-CoV RBD complexed with hACE2 suggests that truncations in the receptor-binding motif (RBM) region of SL-CoV spike protein abolish its hACE2-binding ability [7, 10], and hence the SL-CoV found recently in horseshoe bats is unlikely to be the direct ancestor of human SARS-CoV.
Also, it has been shown that the human SARS-CoV spike protein and its closely related civet SARS-CoV spike protein were not able to use a horseshoe bat (R. pearsoni) ACE2 as a receptor [13], highlighting a critical missing link in the bat-tocivet/human transmission chain of SARSCoV. [Emphasis added [by Fleming].]
However the paper he quoted said that other studies had found that various SARS-like bat viruses were not able to use human ACE2 as a receptor, and next it said that SARS-CoV was not able to use bat ACE2 as a receptor. But the paper he quoted did not say anywhere that SARS-CoV was not able to use human ACE2 as a receptor. [https://pmc.ncbi.nlm.nih.gov/articles/PMC7086629/]
ACE2 binding ability is not even determined by the FCS insert like Fleming suggested but by the RBD, because cleavage by furin occurs at a later step after the virus has already bound to the ACE2 receptor. ChatGPT said that the procedure of viral entry to a human cell consists of these steps:
- Spike protein on virus binds to the ACE2 receptor on human cells via the S1 subunit.
- Conformational change occurs in the spike protein after ACE2 binding.
- Furin cleavage of the spike protein between the S1 and S2 subunits primes the virus for entry.
- Secondary cleavage by TMPRSS2 or cathepsins activates the spike protein, exposing the fusion peptide.
- Membrane fusion is mediated by the S2 subunit, allowing viral RNA to enter the host cell.
- Viral RNA release into the host cell cytoplasm initiates infection.
Fleming wrote: "The furin cleavage site lies in the stable part of the spike protein - the S1 component. All of the mutations being seen in SARSCoV-2 are occurring in the S2 component." He has elsewhere also said that S2 is a more variable region than S1, even though in reality it's of course the other way around.
In Wuhan-Hu-1 S1 is generally considered to extend from amino acid 1
to 685 and S2 is considered to extend from amino acid 686 until the end
of the spike protein. The last R of RRAR is at
position 685.
In NextStrain's subsample of about 5,000 sequences of SARS-CoV-2, about 87% of all amino acid changes in the spike protein were within the range of 1 to 685: [https://docs.nextstrain.org/projects/ncov/en/latest/reference/remote_inputs.html]
$ curl https://data.nextstrain.org/files/ncov/open/global/metadata.tsv.xz|xz -dc>global.tsv
$ sed 1d global.tsv|cut -f57|tr , \\n|grep ^S|sed 's/...//;s/.$//'|awk '{s1+=($0<=685)}END{print s1/NR}'
0.873935
Fleming wrote this about a paper by Zhang et al. which criticized Pradhan's paper:
The investigators concluded that three of the four inserts are present in other coronaviruses:
Among the 4 "insertions" (ISs) of the 2019-nCoV, IS1 has only 1 residue different from the bat coronavirus, and 3 out of 7 residues are identical with MERS-CoV. IS2 and IS3 are all identical to the bat coronavirus. For IS4, although the local sequence alignment by BLAST did not hit the bat coronavirus in Table 4, it has a close evolutionary relation with the bat coronavirus in the MSA. In particular, the first 6 residues in the IS4 fragment "QTQTNSPRRA" from 2019-nCoV are identical to the bat CoV, while the last 4 residues, which were absent in the bat coronavirus or SARS-CoV, have at least 50% identity to MERS-CoV and HCoV-HKU1 34 [Emphasis added [by Fleming].] [34]
Taken together, these statements from research paid for by NIAID (AI134678) and the National Science Foundation (DBI1564756, 1IS1901191) - both agencies involved in the funding of Daszak, Baric, and Zhengli Gain-of-Function research - appear to confirm both Dr. Li-Meng Yan's assertion that SARS-CoV-1 was the first bioweapon and that SARS-CoV-2 is the advanced version noting the PRRA segment.
(Fleming didn't mention that by "the bat coronavirus" the authors he quoted referred to RaTG13.)
I don't understand how Fleming concluded based on the paper he quoted that "SARS-CoV-1 was the first bioweapon". If motifs similar to Pradhan's inserts are missing from SARS-CoV but present in RaTG13, MERS, or HKU1, then how does that show that SARS-CoV was artificially engineered? Was SARS-CoV engineered to remove Pradhan's inserts?
I wrote about Pradhan et al. here: pradhan.html. Pradhan identified the locations of the so-called inserts by doing a pairwise alignment of SARS-CoV-2 against SARS-CoV, so you couldn't tell if the gaps in Pradhan's pairwise alignment were insertions in SARS-CoV-2 or deletions in SARS-CoV. Pradhan's first so-called insert actually looks more like a deletion in SARS-CoV. When I used a more accurate method to determine the positions of the gaps by doing a multiple sequence alignment of SARS-CoV-2 against a diverse set of sarbecoviruses, the gaps near the second and third so-called inserts were placed at different spots than in Pradhan's paper, so it looked like Pradhan's second and third inserts were not inserts at all, but the real second insert was after the motif identified by Pradhan and the real third insert was before the motif identified by Pradhan.
For example in Pradhan's pairwise alignment it looked like SARS-CoV-2
had the insert YYHK relative to SARS-CoV:

But the next image shows that two residues after the
YYHK, SARS-CoV-2 has the motif KSW which is
shared by European and African sarbecoviruses, but after the
KSW SARS-CoV-2 has the insert MESEFR that is
missing from the European and African viruses. So the real insert rather
seems like MESEFR and not YYHK (but before
YYHK SARS-CoV also seems to have an insertion of the
residues SK):
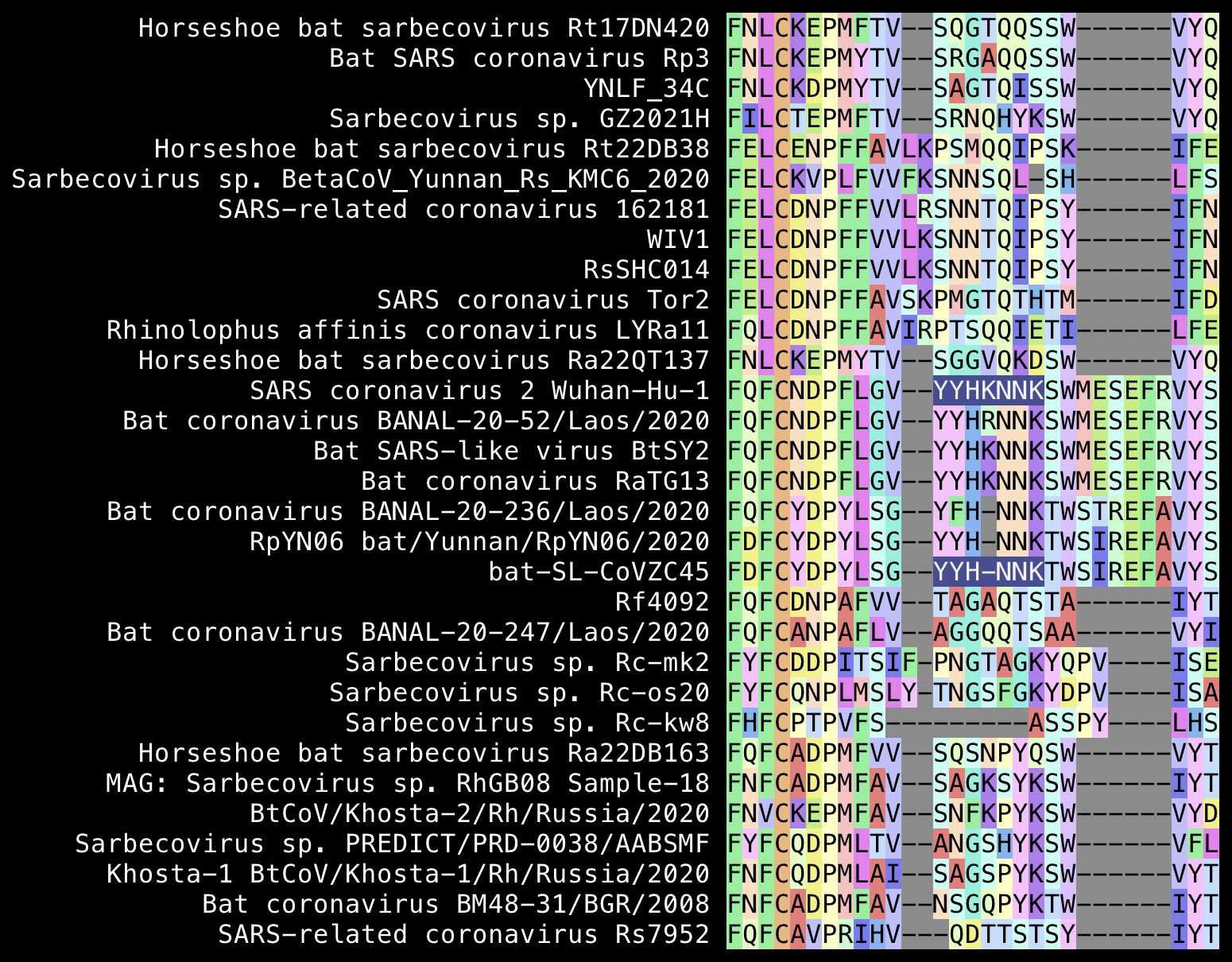
Fleming wrote: "furin (PRRA) cleavage is responsible not only for increasing the infectivity of SARS-CoV-2 but also for converting HIV gp160 to gp120 and gp41".
However the motif cleaved by furin in Wuhan-Hu-1 is RRAR
but the P is just part of the 12-base insert which added
the furin cleavage site.
You can look up the coordinates of features in the envelope protein
of HIV-1 from this spreadsheet:
https://www.hiv.lanl.gov/content/sequence/HIV/MAP/Env_features.xlsx.
It shows that in the HXB2 reference genome of HIV-1 the furin cleavage
motif is REKR:
The Los Alamos National Laboratory has published an HIV subtype
reference collection which contains up to 5 different sequences from
various subtypes of HIV. I downloded envelope protein sequences from the
subtype reference here:
https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html.
I switched the alignment type to "Subtype
reference", changed "DNA/Protein" to
"Protein", and clicked "Get Alignment". In 475 out of 555 sequences the
motif cleaved by furin was REKR, and the next most common
motifs at the same spot of the alignment were RQKR or
RERR (even though I don't know if some other sequences had
a furin cleavage motif at a different spot of the alignment):
$ wget sars2.net/f/HIV1_REF_2023_env_PRO.fasta
$ seqkit seq -s HIV1_REF_2023_env_PRO.fasta|cut -c675-678|sort|uniq -c|sort -r
475 REKR
17 RQKR
14 RERR
9 RGKR
8 RDKR
7 KEKR
5 RSKR
4 RKKR
3 RSRR
3 IEKR
2 RAKR
1 RNKR
1 RHKR
1 RGER
1 REK*
1 KKKK
1 GEKR
1 -EKR
1 ----
Fleming wrote:
Finally, we turn our attention to Luc Montagnier - the discoverer of the Human Immunodeficiency Virus (HIV). Montagnier has published one paper and has submitted another for consideration. In both of these papers, Montagnier utilizes the same BLAST technology for analysis of the genetic code of SARS-CoV-2.
He notes eighteen RNA fragments similar to HIV or simian (higher primates) that have the potential to change the genetic expression of COVID-19:
18 RNA fragments of homology equal or more than 80% with human or simian retroviruses have been found in the COVID-19 genome. These fragments are 18 to 30 nucleotides long and therefore have the potential to modify the gene expression of Covid19. We have named them external Informative Elements or EIE. These EIE are not dispersed randomly, but are concentrated in a small part of the genome.
This is shown schematically in the figure found on page XXX in the insert.
As stated so eloquently by Montagnier, the spike protein not only has the PRRA insertion (twelve nucleotide bases) but also a 1770 nucleotide base (590 amino acid) insertion matching HIV-1:
We have studied the most recent genetic evolution of the COVID-19 strains involved in the world epidemic. We found a significant occurrence of mutations and deletions in the 225 bases area.
On sampling genomes, we show that this 225 bases key region of each genome, rich in EIE, and the 1770 bases SPIKE region evolve much faster than the corresponding whole genome (cases of 44 patients' genomes from WA Seattle state, original epicenter in USA).
In the comparative analysis of both SPIKES genes of COVID-19 and Bat RaTG13 we note two abnormal facts:
- The insertion of 4 contiguous PRRA amino acids in the middle of SPIKE (we show that this site was already an optimal cleavage site BEFORE this insertion).
- An abnormal distribution of synonymous codons in the second half of SPIKE." Finally we show the insertion in this 1770 bases SPIKE region of a significant pair of EIEs from Plasmodium Yoelii and of a possible HIV1 EIE with a crucial Spike mutation.
Fleming seems to have completely misunderstood what Perez meant by the 1,770-base region of the spike protein. It wasn't an HIV-like insertion in SARS-CoV-2 that was missing from other sarbecoviruses, but it was a region where SARS-CoV-2 had high similarity to RaTG13: [https://www.granthaalayahpublication.org/journals/granthaalayah/article/view/IJRG20_B07_3568]

Pezez's 1,770-base region started after the FCS insert and it extended to the end of the spike protein, so it was identical to the S2 subunit of the spike protein. Perez seems to have calculated the length of the region of the wrong, because it should actually be 1,767 or 1,764 nucleotides depending on whether you include the stop codon or not, but Perez made many similar errors in his paper where he gave the wrong coordinates or wrong length for various sequences. But anyway, the black rectangle here shows that there's only 3 amino acid changes in the S2 region, because there's only 3 steps up in the line for nonsynonymous mutations:
In one of his presentations Fleming said: "Luc Montagnier - who was a friend of mine - passed away on the 8th of February, he and Jean-Claude Perez did this work to show the HIV and SIV inserts that they found. PRRA, four proteins, each one has three nucleotide bases. That's 12 nucleotide bases. They found 1,770-nucleotide-base inserts. That's not naturally occurring. They occur one nucleotide base at a time." [https://www.flemingmethod.com/gain-of-function, video from Phoenix, time 43:48] But again Perez's 1,770-base region is not any kind of an insert, but it consists of the entire S2 subunit of the spike protein (plus 3 or 6 extra ghost bases that were likely the due to an error Perez made in calculating the length of the S2 subunit).
I have shown here why Perez's paper is complete junk: pradhan.html#Perez_and_Montagniers_paper_about_BLAST_matches_to_HIV_and_SIV. He used nucleotide BLAST instead of protein BLAST like Pradhan, but he forgot to exclude matches that were on the wrong strand or wrong frame, which will cause the matches to be meaningless because they don't code for the same amino acids in HIV as in SARS-CoV-2. His Table 1 contains 15 BLAST matches to HIV or SIV in SARS-CoV-2, but 10 of the matches are on the wrong strand and 2 of the remaining 5 matches are in the wrong frame:
sseqid evalue nident length pident sstrand SC2_frame HIV_frame right_strand_and_frame JN230738 1.3 16 16 100.000 plus 0 1 no MF373163 0.003 21 21 100.000 minus no JN863831 4.7 17 18 94.444 minus no KR862351 0.11 20 21 95.238 minus no EU875177 0.009 28 32 87.500 minus no JF267434 0.11 18 18 100.000 plus 1 1 yes KJ131112 0.38 19 20 95.000 minus no AF003044 0.38 19 20 95.000 plus 1 1 yes JN091691 0.38 20 22 90.909 minus no HQ644953 0.009 25 28 89.286 plus 0 0 yes L07625 1.3 22 26 84.615 plus 2 1 no JF811228 1.3 16 16 100.000 minus no KC187066 0.009 28 32 87.500 minus no GU481454 0.009 32 39 82.051 minus no LM999945 0.38 25 30 83.333 minus no # sseqid is accession number of HIV or SIV sequence # evalue is number of matches expected to occur by chance for given query and database # nident is number of identical bases within a match # length is length of match in bases # pident is percentage of identical bases (nident/length*100) # sstrand indicates if the subject is on the same strand as the query # SC2_frame indicates coding frame of the match in SARS-CoV-2 # SC2_frame indicates coding frame of the match in HIV or SIV # right_strand_and_frame indicates if sstrand is plus and SC2_frame is same as HIV_frame
(The E-value depends on the size of the BLAST database, but the E-values above were values in a tiny BLAST database that only contained the 15 sequences listed here. In a bigger database the E-values would be much higher, which indicates that all matches are either so short or they have so many errors that they are likely to occur by chance.)
For example the second row of my table above shows that a 21-base segment of SARS-CoV-2 has a perfect to a Swedish HIV sample from 2017. [https://www.ncbi.nlm.nih.gov/nuccore/MF373163] But the matches to SARS-CoV-2 and HIV are on opposite strands so the amino acid translations of the matches are completely different:
SARS-CoV-2: HIV:
<--------------------
--------------------> TACGAAGTCTACTACTGCGTA # reversed version of segment matched in SARS-CoV-2
ATGCGTCATCATCTGAAGCAT ATGCTTCAGATGATGACGCAT # reversed version with complemented bases
AATGCGTCATCATCTGAAGCATTT TATGCTTCAGATGATGACGCATAT
N A S S S E A F Y A S D D D A Y
Perez also claimed to have identified so-called regions A and B in the genome of SARS-CoV-2 that had an unusually high density of BLAST matches to HIV/SIV. However he identified the regions based on the web GUI for BLAST, which had a lot of duplicate results for matches to the same region of SARS-CoV-2 from similar subtypes of HIV. In order to get a more diverse representation of different subtypes of HIV, I tried to instead do a BLAST search of SARS-CoV-2 against sequences from the LANL HIV subtype reference, which consists of 555 sequences from various subtypes of HIV along with a few HIV-1-like sequences of SIV. In the next plot you can only look at the bottom panel where I only included matches to the plus strand (because matches to the minus strand are meaningless because they don't code for the same amino acids), but in the bottom panel Perez's regions A and B didn't get a particularly high density of matches:
In March 2025 Fleming published a document by Soňa Peková which summarized her analysis of DNA contamination in vaccines. [https://www.10letters.org/CzechResearch.pdf] In May 2025 Fleming, Peter Kotlár, and Soňa Peková published a more detailed paper about the analysis. [https://www.fmtvdm.com/_files/ugd/659775_4f73f98b60ae4438bd343c21f2cfc79c.pdf]
Peter Kotlár has the impressive-sounding title of a "plenipotentiary", but it just means that he is a Slovakian MP who was appointed to run a parliamentary commission on COVID. One Slovakian MP said that Kotlár's appointment was a bad joke, and another MP compared it to the recent appointment of a poacher as the head of a Slovakian national park. [https://www.novinky.cz/clanek/zahranicni-evropa-odpurce-ockovani-kotlar-bude-na-slovensku-vysetrovat-management-pandemie-40457747]
Kotlár launched a conspiracy video channel in 2018 with the former mainstream TV host Martina Šimkovičová, but in 2023 both of them managed to get elected to the Slovakian parliament. Videos by Kotlár's commission were posted on Šimkovičová's Facebook page, but they were later made private. [https://www.facebook.com/martina.peter.111/videos/] The only Western people who did video presentations for the commission were Richard Fleming and Charles Rixey:
In 2020 Soňa Peková was reported to face up to 10 years in prison for medical insurance fraud. [https://archiv.hn.cz/c1-66808150-lekarka-pekova-pujde-s-janouskem-pred-soud-kvuli-machinacim-s-pojistovnami-skoda-podle-zaloby-cini-240-milionu] She was indicted in 2022 but apparently she did not serve time in jail. [https://ct24.ceskatelevize.cz/clanek/domaci/soud-zprostil-obzaloby-lobbistu-janouska-i-dalsi-ctyri-lidi-v-kauze-spolecnosti-chambon-23287]
By 2023 she had become a member of a neo-Theosophical cult called ALLATRA. [https://x.com/search?q=So%C5%88a+Pekov%C3%A1+allatra+until%3A2024-7-1&f=live] In 2023 Peková presented genetic evidence that Slavs are the oldest ethnic group after the fall of Atlantis. [https://cz24.news/video-molekularni-geneticka-mudr-pekova-o-genetice-nasich-predku-slovanu-jako-nejstarsim-etniku-po-padu-atlantidy-sile-a-vyjimecnosti-slovanu/] In a video in 2024, Soňa Peková talked about the Pleiadians and how contact with aliens is a win-win situation, and she also said that Gaia has the capacity to reach up to 9D reality and not only 5D, even though it's possible that vaccinated people cannot move to a higher dimension. [https://www.podtatransky-kurier.sk/galaxia/sona-pekova-gaia-ma-byt-az-9d]
ALLATRA says that the world will end in the year 2036, they promote theories about ancient aliens and 5D ascension, and they say that reality is contaminated by Draconian reptilians who have a goal of enslaving humanity. [https://infosecurity.sk/zahranicne/sona-pekova-preslavila-sa-dezinformaciami-o-covid-19-v-sucasnosti-ma-blizko-k-ezoterickej-sekte/] A member of ALLATRA called Zhanna is allegedly an Anunnaki who makes TikTok videos from a space station. The primary book of ALLATRA is presented as a dialogue between the author of the book and the ascended master Rigden Djappo, who is a mythical character invented by the Russian Theosophist Nikolas Roerich. [https://handwiki.org/wiki/Organization:AllatRa]
ALLATRA is overtly based in Ukraine, but they run a multilingual media operation that produces conspiracy content in various languages, and much of their content is initially released in English but then translated to various Slavic languages. It is reminiscent of the multilingual empires operated by the Unification Church, Falun Gong, and Miles Guo's cult. All of them are based in New York even though they primarily target East Asian countries, and the leaders of all three cults moved from an East Asian country to New York to run the cult. But ALLATRA seems very much like a version of Falun Gong that was created for the Eastern European demographic instead of China.
ALLATRA's international representative is Egon Cholakian, who claims that he is an intelligence expert who studied under Henry Kissinger, and that he had a close working relationship with the head of the CIA while he resided in Iran, and that he was a founding member of the OSINT Foundation. [https://earthsavesciencecollaborative.com] However his bio seems to be mostly fake and I didn't find it corroborated by outside sources.
Egon Cholakian's website used to have an AI-generated photo where he posed together with the Nobel laureate Robert Merton, but if you zoomed in on the background, there were photos on display where people were missing faces, and the backs of books on a bookshelf didn't have any legible text. [http://web.archive.org/web/20240717153504/earthsavesciencecollaborative.com/assets/slider/13.jpg?t=1]
ALLATRA has a YouTube channel that consists of AI-generated videos that employ the avatar of Egon Cholakian:
In one of the videos Egon Cholakian said that "The claim by Russian media that I am a deep fake is so absurd it would be laughable if it weren't part of a larger coordinated effort to mislead you." [https://www.youtube.com/watch?v=J_EM0Dp2Cmg&start=1m46s] In another video he called himself a "specialist in intelligence and disinformation". [https://www.youtube.com/watch?v=Q2GVN5Hj16Y&t=18m30s] In the same video he also said that Scientology was an "international pro-democratic organization", and that Scientology was unjustly persecuted in Russia. [time 1:32:47]
ALLATRA claims that they are not a cult, but they made an 8-hour documentary about how the anti-cult movement is the root of all the world's evils, and how the anti-cult movement is the power behind Putin's throne, and the anti-cult movement did 9/11, and the anti-cult movement is secretly orchestrating school shootings. [https://www.youtube.com/watch?v=4l4CAqW1B2g&t=21060s] They film also included a long segment about how Falun Gong was persecuted in China.
The secular arm of ALLATRA is known as the Creative Society, which ALLATRA aims to one day make into the governing body of the world. Their emblem looks similar to the emblem of the Theosophical Society, except the head of the snake has been replaced with an arrowhead, the star of David has been replaced with a triangle, the ankh has been replaced with a Moebius strip, and the swastika has been removed:

In the same way Scientology is allied with the Nation of Islam, ALLATRA is allied with an African American freemasonic lodge called the World's Masonic United Nations. The Facebook page of Creative Society featured a speech by the Most Puissant Sovereign Grand Commander of the World's Masonic United Nations, who said that the world's strongest foundation is the Eight Pillars of Creative Society, and he said that "we support the Creative Society project and we will use all legal and peaceful means to implement this ideal as broadly and quickly as possible": [https://www.facebook.com/watch/?v=264451399321129]
In a Czech-language Facebook post, Soňa Peková wrote that humans have to find peace within themselves, "So that we can start building a new society as quickly as possible, based on the principles of humanity, solidarity, generosity, equality and belonging." [https://www.facebook.com/story.php?story_fbid=122184364922255070&id=61557652117076] She used the same Czech word for "society" that is used in the Czech name for Creative Society. So by building a new society, she might have referred to the Creative Society, which is the organization that ALLATRA aims to establish as the governing body of the world, so that one day the world will be ruled by ALLATRA. [https://creativesociety.com/about-the-project]
Similar to Scientology and NOI, ALLATRA is also concerned with the topic of autism, except ALLATRA says that autism is caused by microplastics and not by vaccines. [https://www.facebook.com/CreativeSociety.en/videos/1206675507639876]
The Creative Society has developed a "comprehensive mathematical and tectonophysical model of contemporary changes occurring on earth". [https://creativesociety.com/climate-model] The term "earth changes" was coined by the Theosophical guru Edgar Cacey.
The Facebook page of Creative Society has a video titled "Trailer. Who Has a Sharp Mind?" Its description says: "Who possesses a sharp mind and an innate organizational talent? Whose communication skills enable them to always maintain good relations with everyone? Who can find a way out of any seemingly hopeless situation? This is not just a skill, but a talent that is passed on genetically. People of Israel, we consider ourselves the chosen people." [https://www.facebook.com/CreativeSociety.en/videos/6980783995281874]
The Slovak Academy of Sciences published the following statement about the paper by Richard Fleming, Peter Kotlár, and Soňa Peková: [https://www.sav.sk/?lang=sk&doc=services-news&source_no=20&news_no=12787, translated]
The Slovak Academy of Sciences is responding to a publication published in the HSOA Journal of Angiology and Vascular Surgery, co-authored by the Slovak Government's Plenipotentiary for Review of the Resource Governance and Management Process during the COVID-19 pandemic, Peter Kotlár.
The Slovak Academy of Sciences is following with interest the attempts to re-analyze vaccines that have been analyzed many times. However, the published publication does not adequately describe the methods, lacks controls, and does not clearly define the input quantities of material. The published claims are not confronted with existing knowledge or other methods, which is necessary given the seriousness of the findings presented.
The title of the publication suggests a quantitative analysis, but the results are not compared to the prescribed and strictly controlled quantitative limits of residual DNA, and estimated numbers of molecules are given, which can be misleading. Strong conclusions are not supported by sufficiently strong data.
The scientific shortcomings of the aforementioned publication reflect the fact that the journal in which it was published is classified as a predatory journal. This particular journal does not meet even basic quality criteria, is not registered in recognized databases, has a very short review process and unclear review procedure, and does not have defined basic rules for presenting results and methodologies.
The Slovak Academy of Sciences strictly condemns publishing in predatory journals and warns that publications published in this way do not bring value and are of no importance to the scientific community, medical practice, or the public.
The paper by Fleming, Kotlár, and Pekova was published in a journal of the predatory publisher Herald Scholarly Open Access. [https://www.heraldopenaccess.us/openaccess/quantitative-analysis-of-nucleic-acid-content-in-spikevax-moderna-and-bnt162b2-pfizer-covid-19-vaccine-lots] One of their journals published a paper by Hellsing et al. about the benefit of garlic in fighting vampires, which said the purpose of the paper was "improving the stature of vampire hunting among the scientifically literate populace, and testing whether or not AIMS journals are predatory". [https://www.heraldopenaccess.us/openaccess/allium-argenti-et-aqua-sancta-transgressing-molecular-boundaries-in-hematology-post-alucard]

{kind=link}